 電子發燒友App
電子發燒友App


作者:吳敬華 1,2 楊菁 1劉高瞻 1王脂胭 1,2張秩華 1俞海龍 2,3姚霞銀 1,2 黃學杰 2,3
摘 要 采用固體電解質取代液態有機電解液的固態鋰電池,有望使用更高比容量的正、負極材料,從而實現更高比能量的電池體系,同時可徹底解決電池的安全性問題,符合未來二次電池發展的方向,是電動汽車和規模化儲能的理想電源。為了實現兼具高比能量、高安全性、長壽命等特性的固態電池,進而推進全固態鋰電池的實用化,2011—2021年間各國的科學家做了大量工作,并取得了許多突破性進展。本文以固態鋰電池關鍵材料為出發點,回顧了2011—2021年以來固態電池的研究進展,包括鋰離子固體電解質材料,電極/電解質界面調控,固態電池技術等方面,總結了現在存在的挑戰及解決方案,并對該領域未來可能的發展提出了展望。
作為清潔能源的代表,鋰離子電池由于其高比能量/功率、環境友好以及使用壽命長等特點,成為最具競爭力的電化學儲能器件之一。目前,鋰離子電池在便攜式電子設備和電動汽車上已經得到廣泛應用。但是,基于氧化物正極與石墨負極的傳統鋰離子電池的能量密度越來越接近其理論上限,同時,由于采用有機液態電解液,鋰離子電池在充放電過程中不可避免地發生副反應,以及電池循環過程中電解液揮發、泄漏等現象均會導致電池容量的不可逆衰減,影響鋰離子電池的使用壽命。此外,由有機易燃電解液引起的安全問題,引發民眾對鋰離子電池安全性的疑慮,尤其在一些關鍵行業,如航空航天、電動汽車、儲能電網等領域,電池的安全性顯得至關重要。
采用固態電解質取代液態有機電解液的固態電池,有望同時解決傳統鋰離子電池面臨的比能量、循環壽命以及安全性等困境,符合未來大容量二次電池發展的方向,是電動汽車和規模化儲能的理想電源。與傳統液態鋰離子電池相比,全固態鋰電池具有如下優勢:①消除液態電解液泄漏和腐蝕的隱患,熱穩定性更高;②穩定且較寬的電化學窗口,可匹配高電壓正極材料;③固態電解質一般為單離子導體,副反應少,循環壽命更長;④全固態鋰離子電池可通過多層堆垛技術實現內部串聯,獲得更高的輸出電壓。因此,全固態鋰電池被認為是鋰電池的終極目標。
作為固態鋰電池核心組成部分——固體電解質是實現固態鋰電池高能量密度、高循環穩定性和高安全性能的關鍵。固體電解質又稱快離子導體,主要包括聚合物固體電解質和無機固體電解質兩大類。其中無機固體電解質又包括:硫化物固體電解質,氧化物固體電解質,硼氫化物固體電解質以及鹵化物固體電解質等。無論采用何種固體電解質,由此帶來的界面問題對于電池性能的影響都至關重要。在全固態鋰電池中,電極與電解質之間的界面接觸由固液面接觸變為固固點接觸,由于固相無潤濕性,因此固固界面將形成更高的界面電阻。同時,固體電解質,尤其陶瓷電解質中有大量的晶界存在,且晶界電阻往往高于材料本體電阻,不利于鋰離子在正負極之間傳輸。
為了推進全固態鋰電池的實用化,最近十年各國的科學家都做了大量工作,并取得了許多突破性進展。本文主要回顧了2012—2022年以來國內外在電解質材料、電極/電解質界面以及固態電池技術等方面的研究進展,并針對當前在固態電池研究中面臨的困難和挑戰,總結了推進固態電池實用化過程中的常用策略,最后探討了全固態鋰電池可能的研究方向和發展趨勢。并以此文祝賀《儲能科學與技術》創刊十周年。
1 氧化物固體電解質進展
氧化物固體電解質材料具有安全性能高、穩定性良好、成本低廉、環境友好等優點,是儲能應用的研究熱點。氧化物固體電解質主要包括NASICON(sodium superionic conductor)型結構氧化物電解質、石榴石結構氧化物電解質和鈣鈦礦結構氧化物電解質。近十年來針對氧化物固體電解質,主要開展了其制備、改性、應用等方面的研究。
1.1 NASICON型結構固體電解質
NASICON型結構固體電解質的通式為Li[A2B3O12],其中A、B分別代表四價和五價骨架離子。NASICON型結構固體電解質制備工藝簡便,易于加工處理,對空氣穩定,熱穩定性和力學性能良好,是一類重要的氧化物固體電解質材料。常見的NASICON型結構固體電解質根據化學組成可分為LiZr2(PO4)3(LZP),LiTi2(PO4)3(LTP)和LiGe2(PO4)3(LGP)。其中,LTP和LGP的離子電導率明顯高于LZP,為近年來主要研究的NASICON型氧化物固體電解質體系。為提高LTP和LGP固體電解質的離子電導率,最常用的手段是通過離子取代來調控離子傳輸通道。在LTP和LGP中,采用Sc、Al、Y、Ga等對Ti或Ge進行部分取代,以及采用Si對P進行部分取代均可有效提高離子電導率。其中,Al3+取代被證明是最有效的手段。因此在LTP和LGP體系中,Li1.3Al0.3Ti1.7(PO4)3(LATP)和Li1.5Al0.5Ge1.5(PO4)3(LAGP)具有很高的離子電導率,兩者電導率均可達到約10-3 S/cm量級,是當前最常見的研究體系。除了傳統合成方法之外,Zhu等提出了一種利用NASICON骨架結構制備高性能固態電解質的方法。研究者首先制備了NASICON結構的Na3Zr2Si2PO12(NZSP)前驅體,然后通過鋰鈉離子交換獲得了同樣具有NASICON結構的Li3Zr2Si2PO12(LZSP)電解質。該方法的優點在于在保留鈉超離子導體框架結構的同時,引入了鋰離子并穩定在低配位結構中,獲得了較大的鋰離子傳輸路徑,從而確保了其高離子電導率。合成的LZSP在室溫下的離子電導率達3.59×10-3 S/cm,活化能為0.21 eV。此外,LZSP繼承了NZSP優異的空氣穩定性,而且應用于固態電池時表現出優異的對鋰穩定性、循環穩定性和倍率性能。
雖然LAGP和LATP電解質具有離子電導率高的優點,但由于內部存在的Ge4+和Ti4+在熱力學上對鋰不穩定,極易被鋰還原。為了提高其實用性,現階段的研究主要通過組分優化以及引入界面層等方式來改善其負極穩定性。效果較好的界面層材料包括非晶材料、氟化物材料、氮化物材料、氧化物材料和其他固體電解質材料。不同的界面修飾層材料對比于表1中列出。Hao等報道通過在LATP固體電解質表面濺射ZnO層,構建具有低電子電導和良好潤濕性的Zn@Li2O界面層,有效改善了LATP/Li的界面穩定性(圖1)。Liu等通過ALD手段在LATP固體電解質表面包覆Al2O3層,包覆后的LATP電解質應用在鋰對稱電池中可實現600小時穩定循環。在固體電解質表面包覆PEO、LiPON等電解質材料來穩定NASICON型結構固體電解質與金屬鋰的界面,也是一種有效的方法。另外,鑒于非晶材料具有良好的對鋰穩定性,制備對鋰穩定的非晶NASICON型結構固體電解質成為一種改善界面穩定性的方法。2019年,Zhang等通過旋涂的方式,在LAGP玻璃陶瓷片上均勻涂覆了一層對鋰穩定的非晶LAGP層。采用涂覆后LAGP電解質組裝的固態電池在容量和循環穩定性上均遠優于未涂覆電解質組裝的固態電池。
表1 NASICON/Li界面不同的界面修飾層材料對比

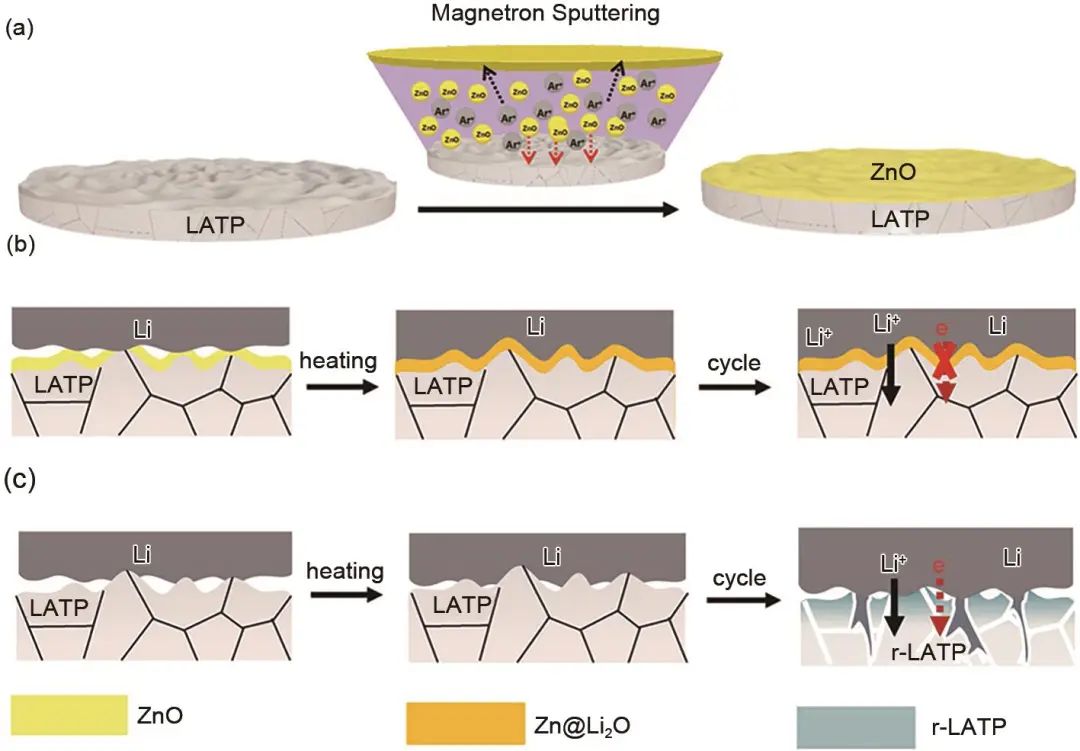
圖1 (a) 磁控濺射在LATP表面制備超薄ZnO2示意圖;(b) 無ZnO層時鋰金屬負極與LATP界面變化;(c) 有ZnO層時鋰金屬負極與LATP界面變化
1.2 石榴石結構固體電解質
石榴石結構固體電解質Li7La3Zr2O12(LLZO)具有良好的離子傳輸性能(10-3 S/cm)。除具有氧化物固體電解質一貫的穩定性優點外,還表現出遠優于其他種類氧化物固體電解質的對鋰金屬穩定性,因此其在固態電池應用中極具潛力。LLZO存在立方相(c-LLZO)和四方相(t-LLZO)兩種晶體結構。其中,c-LLZO在室溫下是亞穩定相,離子電導率更高;而t-LLZO屬于室溫穩定相,離子電導率低。設法在室溫下穩定立方相,以提高其離子電導率是當前LLZO材料制備領域的研究重點。另外,雖然LLZO對鋰金屬穩定,但內部存在的缺陷依然會導致枝晶的生長,最終導致固態電池失效,因而提高LLZO致密度也是當前重要的研究方向。在采用LLZO材料組裝固態電池時,其與鋰金屬負極良好的界面接觸對鋰離子在界面的均勻傳輸極為關鍵。由于LLZO與鋰金屬接觸屬于點對點的固-固接觸,接觸面積小,且兩者界面能不匹配使得不能實現潤濕,會導致界面阻抗較大和界面處鋰枝晶的生長。因此,通過減小LLZO與鋰金屬之間界面能的差別,改善兩者間潤濕性,優化界面接觸,是實現LLZO應用的重要前提。
采用合適離子取代骨架離子是促進c-LLZO在室溫下穩定和提高離子電導率的有效策略。Buschmann等通過對LLZO的合成,證實了Al摻雜有利于立方相結構的穩定和離子電導率的提高。后續研究中,研究人員發現除Al3+外,還有其他合適取代離子也可用來穩定結構、降低燒結溫度、抑制雜相生成和提高離子電導率。目前已經進行的取代包括采用Sr、Y和Ca取代La和采用Nb、Ti、Ta、Sb、W、Te等取代Zr。研究表明,離子取代量對LLZO離子電導率的提升存在影響:取代量過低時無法穩定高電導率立方相,而取代量過高時無法被容納在晶體結構框架中導致產生低電導率雜相。此外,致密的LLZO固體電解質內部電流密度均勻,有助于鋰金屬的均勻沉積從而抑制枝晶生長。對于LLZO致密度的提高,研究人員主要采用的手段包括優化制備方法、助熔劑復合和單晶材料制備,最常見的優化制備方法是熱壓法。2016年,Sharafi等利用快速熱壓技術強化制備了相對密度達到(97±1)%的Li7La3Zr2O12固體電解質材料。對于助熔劑復合法,低熔點助熔劑如MgO、LiBr、LiBO2等被應用于致密LLZO固體電解質的制備,可有效提高材料的致密度,還可在一定程度上實現良好的晶粒間接觸,進而提高離子電導率。而LLZO單晶制備,是近年來出現的一種致密度提高的新方法。Kataoka等通過浮區熔融生長法首次成功生長出厘米級單晶Li6.5La3Zr1.5Nb0.5O12和Li6.5La3Zr1.5Ta0.5O12,獲得了極高的鋰離子電導率(1.39×10-3 S/cm和1.27×10-3 S/cm)。得益于單晶材料極高的致密度,采用Li6.5La3Zr1.5Nb0.5O12電解質片組裝的對稱電池,表現出良好穩定性。為實現LLZO固體電解質與鋰金屬之間的潤濕,潤濕界面層被廣泛應用,進而改善固-固界面接觸、抑制鋰枝晶生長以及緩沖電池循環過程中材料的體積變化。常見潤濕層材料包括金屬/非金屬單質、氧化物、硫化物、氮化物、氟化物、聚合物等。不同潤濕層材料對比如表2所示。Han等報道通過在LLZO表面沉積一層5 nm厚度的Al2O3,改善了對鋰潤濕性,消除了界面的空隙。同時,由于Al2O3與鋰發生原位反應可在界面生成離子導電的親鋰層,有效抑制了雜質和界面缺陷的生成。在多種因素作用下,界面阻抗由1710 Ω·cm2降至34 Ω·cm2。Luo等通過CVD手段在LLZO表面鍍上一層超薄的非晶Si層(見圖2),可將鋰與LLZO界面阻抗由925 Ω·cm2降至 127 Ω·cm2,實現了將原先疏鋰的表面轉變為親鋰表面。
表2 LLZO/Li界面不同的潤濕層材料對比
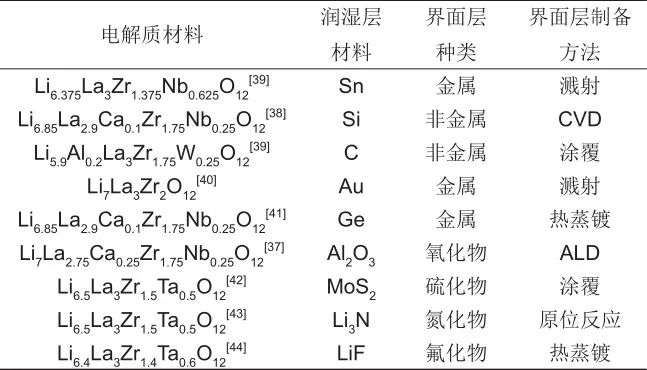
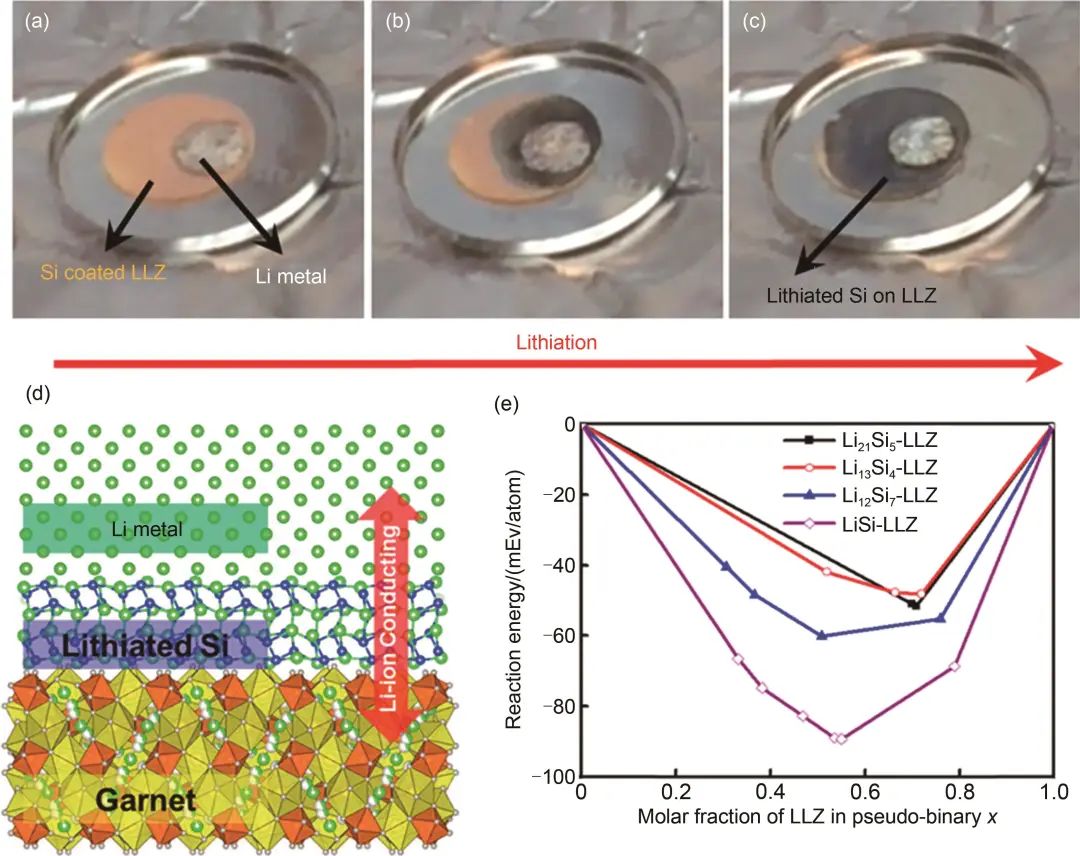
圖2 具有Si鍍層的LLZO與熔融鋰的反應變化趨勢(a) 反應前照片;(b)、(c) 反應后照片;(d) 原位反應生成親鋰層示意圖;(e) 原位反應生成親鋰層與LLZO和鋰的反應能量計算結果
1.3 鈣鈦礦結構固體電解質
鈣鈦礦結構固體電解質的通式為Li3xLa2/3-xTiO3(LLTO)。鈣鈦礦結構固體電解質電導率較高,熱穩定性和力學性能良好,但制備溫度較高。為提高LLTO固體電解質的離子電導率,通常采用的策略包括離子取代、復合和燒結氣氛調控。研究表明,采用Pr、Nd和Y等對LLTO中的La進行部分取代,或者采用Al、Zr、Cr、Nb、Ta、W等對LLTO中的Ti進行部分取代,可有效提高離子電導率。值得注意的是,對La位取代來說,取代離子的半徑越大,鋰離子傳輸通道瓶頸尺寸越大,越能提高離子電導率。相反,對Ti位取代來說,取代離子的半徑越小,使Ti—O原子間距離縮短,Ti—O鍵強度得到增強,越有利于提高離子電導率。與LLTO較高的晶粒電導率相比,LLTO的晶界電導率一般要低2~3個數量級,是LLTO離子電導率提高的重點方向。通過復合LiF等增加鋰離子濃度,或復合鋰硼氧化物(Li2O-B2O3)等低熔點助劑來改善燒結性能、提高致密度,均被證明可提高晶界電導率。此外,通過排除燒結氣氛中的水和CO2,避免低電導Li2CO3的生成也有利于離子電導率的提高。
與NASICON型結構固體電解質類似,LLTO固體電解質由于存在易被還原的Ti4+,也存在對鋰界面穩定性問題。Yan等提出采用混合離子/電子導體層來解決穩定性問題。研究中以甲苯為催化劑,引發了LLTO與金屬鋰的反應,形成了Li0.35La0.52[V]0.13TiO3混合離子/電子導體層。該混合離子/電子導體層緩沖了鋰離子濃度梯度,并使鋰金屬表面的二次電流均勻分布,有效抑制了副反應發生和枝晶生長。目前,LLTO的主要合成方法都需要較高的燒結溫度,容易導致鋰的揮發和晶界阻抗的提高。因此,降低燒結溫度,并降低晶界阻抗將是未來LLTO固體電解質研究的重點。
2 硫化物固體電解質進展
硫化物固體電解質在室溫下具有較高的離子電導率,范圍從10-4到10-2 S/cm。同時,硫化物固體電解質具有可忽略的電子電導率和良好的力學性能,有利于全固態電池中電極/電解質形成良好的固-固接觸界面,從而優化全固態電池的循環穩定性。因此,許多工作從硫化物固體電解質體系入手,致力于開發出具有高離子電導率,寬電化學窗口,具有良好化學穩定性和空氣穩定性的理想固體電解質材料。圖3整理了2011—2021年硫化物固體電解質研究中具有代表性的工作,以下將按照Li-P-S二元硫化物固體電解質,Li10GeP2S12,Li6PS5X(X=Cl,Br,I)三個體系分類分別介紹硫化物固體電解質近十年工作進展。
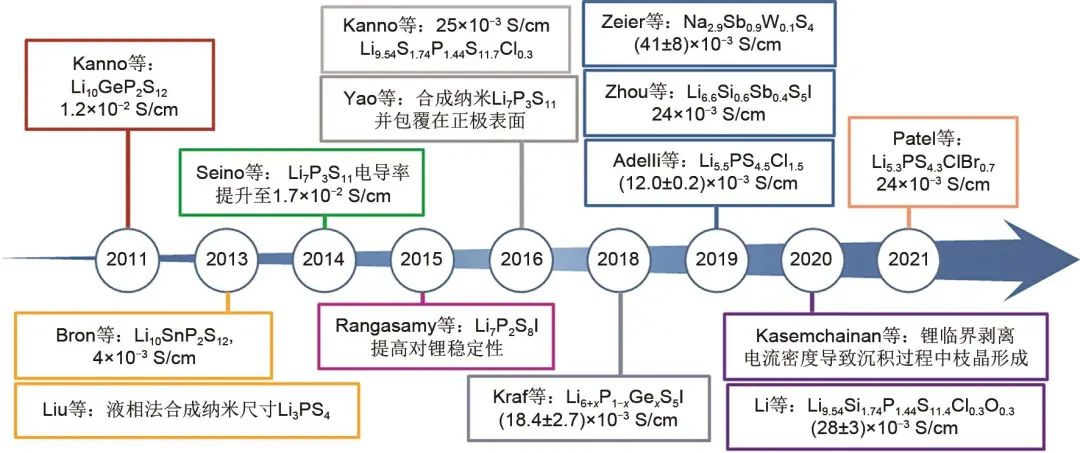
圖3 近十年硫化物固體電解質重要進展
2.1 Li-P-S體系
Li2S-P2S5體系硫化物固體電解質的研究始于1981年,近年來廣受關注的是具有良好對鋰穩定性和對空氣穩定性的75Li2S-25P2S5(Li3PS4)體系材料和電導率較高的70Li2S-30P2S5(Li7P3S11)體系材料。
早期Li3PS4電解質材料主要通過固相法合成,顆粒尺寸較大,電導率較低。Liu等采用液相法合成了具有納米孔隙結構的Li3PS4,將電解質材料尺寸降低到納米級,穩定了高電導率的β相,同時使電解質材料比表面積增加,提高了表面電導性能,制備得到的Li3PS4電解質室溫離子電導率為1.6×10-4 S/cm。除了室溫離子電導率和顆粒尺寸,近年來針對Li3PS4對鋰穩定性改性的研究也受到廣泛關注。Rangasamy等通過固相法在Li3PS4中摻雜LiI,合成了Li7P2S8I玻璃陶瓷電解質,具有6.3×10-4 S/cm的室溫離子電導,同時,含I相的存在使Li7P2S8I電解質的對鋰穩定性提高。
與Li3PS4電解質相比,Li7P3S11電解質具有更高的室溫離子電導率。2014年,Seino等通過調整熱處理條件,降低了固體電解質晶界阻抗及電解質體相空隙對電解質離子電導率的影響,將Li7P3S11電解質的室溫離子電導率進一步提升至1.7×10-2 S/cm。在電解質顆粒尺寸控制方面,Yao等通過液相法合成Li7P3S11電解質,其顆粒尺寸為400 nm~1 μm,同時具有1.5×10-3 S/cm的高室溫離子電導率,并通過液相法將電解質包覆在Co9S8電極材料表面,獲得了更連續的鋰離子傳輸通道,從而使全固態電池實現更好的電化學性能。
2.2 Li11-xM2-xP1+xS12 (M = Ge, Sn, Si) 體系
為了進一步優化Li2S-P2S5的離子電導率及其他電化學性能,許多工作通過引入GeS2、SiS2、SnS2或Al2S3等第三組分獲得三元體系。其中Li2S-GeS2-P2S5具有很高的離子電導率,是三元硫化物電解質中研究最多的體系。2011年日本東京工業大學的Kamaya等制備出Li10GeP2S12電解質,其電導率高達1.2×10-2 S/cm,是第一種離子電導率可與液態電解液相媲美的固體電解質。Li10GeP2S12電解質在較低的溫度下也表現出高離子導電性,在-30 ℃下為1.0×10-3 S/cm,-45 ℃下為4.0×10-4 S/cm。雖然Li10GeP2S12具有較高的離子電導率,但Ge元素的高成本和對鋰金屬的不穩定性限制了其在全固態鋰電池中的大規模應用。因此,許多具有成本優勢的元素被用來取代Ge元素。為了降低電解質的成本,同時保證電解質高室溫離子電導率,Kato等于2016年通過采用Si、Cl同時取代Ge和S,制備出Li9.54Si1.74P1.44S11.7Cl0.3電解質,其晶體結構與Li10GeP2S12電解質相似,具有25×10-3 S/cm的超高室溫離子電導率。在此基礎上,Li等通過O取代具有Li10GeP2S12構型的Li9.54Si1.74P1.44S11.7Cl0.3中的S,合成了Li9.54Si1.74P1.44S11.7-zCl0.3Oz固態電解質材料(0
2.3 Li6PS5X(X=Cl, Br, I)體系
硫銀鍺礦相Li6PS5X(X=Cl, Br, I)電解質是具有另一種晶體結構的硫化物固體電解質,由具有較高Ag+導電性的Ag8GeS6演變得到。以Li替代Ag元素,P替代Ge元素,得到Li7PS6,硫銀鍺礦鋰型化合物具有較高的鋰離子電導率。然而,Li7PS6在高溫下為立方相,在低溫下會轉變為正交晶相,溫度穩定性較差。2008年,Deiseroth等以鹵素陰離子部分取代S2-離子,首次得到通式為Li6PS5X(X=Cl,Br,I)的硫銀鍺礦鋰型化合物。相比于Li10GeP2S12電解質,硫銀鍺礦相具有更加優良的化學和電化學穩定性,因此引起眾多研究者關注,成為最近幾年的研究熱點。
對硫銀鍺礦相Li6PS5X(X=Cl,Br,I)電解質來說,鹵素位元素對其晶體結構和室溫離子電導率有較大影響,當鹵素位X=Cl或Br時,由于Cl-或Br-離子半徑小,S2-和X -排列具有一定的無序性,便于鋰離子傳導,其室溫離子電導率約10-3 S/cm;當鹵素位為I時,晶體結構中S2-和X -有序排列,室溫離子電導率約10-5 S/cm。由此可見,X -與S2-排列無序度對鋰離子在晶格中的擴散具有重要的影響。Adeli等報道了一種富含鹵素的硫銀鍺礦類固體電解質Li6-xPS5-xCl1+x,通過單價的Cl-部分取代二價的S2-,晶體結構中無序位點和鋰空位增多,減弱了游離鋰離子同周圍骨架的相互作用,因此可使鋰離子擴散系數得到提高。冷壓下,Li5.5PS4.5Cl1.5在298 K時的離子電導率為(9.4±0.1)×10-3 S/cm,燒結后為(12.0±0.2)×10-3 S/cm。基于富含鹵素的硫銀鍺礦相電解質,Patel等報道了Li6-xPS5-xClBrx(0≤x≤0.8)電解質,其中Li5.3PS4.3ClBr0.7在25 ℃下有24×10-3 S/cm的高離子電導率,以及0.155 eV的極低鋰遷移勢壘。引入溴原子后,Li6-xPS5-xClBrx(0≤x≤0.7)仍保持Li6PS5Cl結構。Cl-、Br-與S2-共同占位在4a/4d,且在x接近0.7時產生了類液相的鋰亞晶格結構,增強了鋰離子遷移能力。此外,陰離子種類的多樣化以及鋰空位的產生誘發了Li6-xPS5-xClBrx中過配位與配位熵的產生,這些也都增強了其鋰離子傳輸能力。
通過X -與S2-排列無序度調控,Li6PS5I基硫銀鍺礦相電解質的室溫離子電導率也有大幅提高。2018年,Kraft等通過X射線和中子衍射以及阻抗和核磁共振譜研究了異價替換對電解質Li6+xP1-xGexS5I性能的影響。研究表明,隨著Ge含量的提高,材料表現出陰離子位點的無序化,離子運動的活化能明顯下降,使得該材料獲得了當時硫銀鍺礦類電解質中最高的鋰離子電導率,冷壓狀態下為(5.4±0.8)×10-3 S/cm,燒結狀態下為(18.4±2.7)×10-3 S/cm。Zhou等人同樣通過P位異價元素取代合成Li6.6Si0.6Sb0.4S5I電解質,其冷壓壓片后具有14.8×10-3 S/cm的高離子電導率,而燒結片的離子電導率能達到24×10-3 S/cm。同時,該電解質具有極佳的低溫離子電導性能,在-78 ℃下仍具有0.25×10-3 S/cm的離子電導率。該電解質與金屬鋰具有良好的穩定性,用其組裝的對稱電池在0.6 mA/cm2的電流密度下,可以穩定循環1000 h。
除了離子電導率的提高,針對電解質層中鋰枝晶生長的問題,Kasemchainan等使用三電極電池裝置[圖4(a)]對Li-Li對稱電池進行表征,可以區分鋰嵌入和鋰剝離過程。測試發現,對稱電池臨界電流密度受臨界剝離電流密度影響較大,增大壓力可以提高電池臨界電流密度。如圖4(b)所示,當施加電流密度大于臨界剝離電流密度時,剝離過程中鋰入不敷出,金屬鋰/電解質界面出現孔洞,在沉積過程中,金屬鋰填充部分孔洞,且鋰枝晶沿電解質片內的空隙生長。隨著循環進行,界面處空洞越來越多,導致剝離過程中界面接觸性能差,局部電流密度增大,循環惡化,枝晶生長加速,進而失效短路。
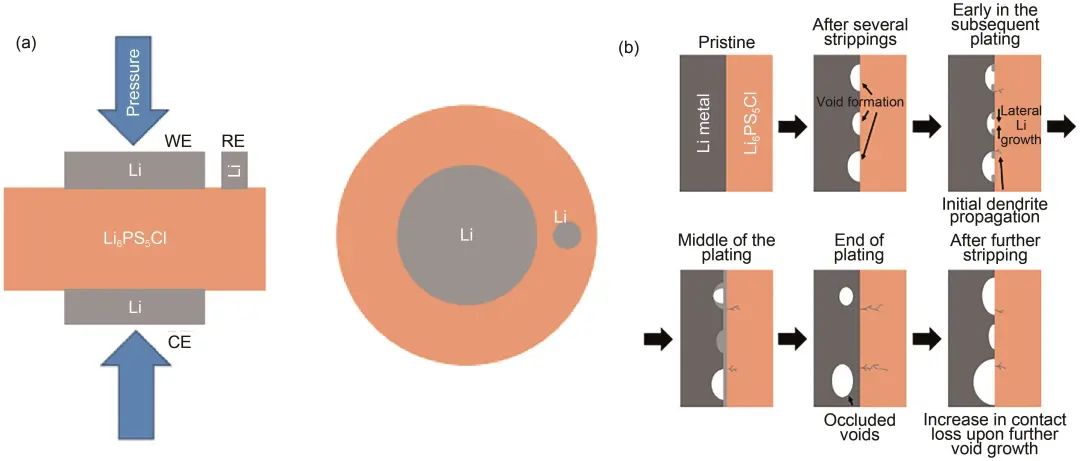
圖4 (a) 三電極電池示意圖(WE,工作電極;RE,參比電極;CE,對電極);(b) 鋰/Li6PS5Cl界面在總電流密度高于臨界剝離電流密度情況下循環機理示意圖
在硫化物鈉快離子導體方面,Fuchs等報道了Na2.9Sb0.9W0.1S4硫化物鈉快離子導體,其室溫離子電導率為(41±8)×10-3 S/cm,是目前電導率最高的硫化物固體電解質。
3 聚合物固體電解質進展
3.1 聚合物固體電解質簡介
相較于無機固體電解質,溶解鋰鹽的固體聚合物電解質(SPE)具有柔韌性好、質量輕、成本低以及易于加工等優勢。在各種聚合物基體如聚環氧乙烷(PEO)、聚丙烯腈(PAN)、聚偏氟乙烯(PVDF)、聚(偏二氟乙烯-六氟丙烯)(PVDF-HFP)和聚甲基丙烯酸甲酯(PMMA)中,PEO含有乙氧基鏈段,對鋰鹽具有高的溶解能力,是研究最為廣泛的聚合物基體材料。早在1973年,Fenton等率先發現部分堿金屬鹽可以和PEO形成聚合物和鹽的絡合物,并對其離子導電性進行了研究。但早期制備的PEO基聚合物電解質的室溫離子電導率僅僅只有10-7~10-5 S/cm,這主要由于PEO是一種易于結晶的聚合物基體,在較低的溫度下PEO基聚合物電解質內僅含有極其少量的無定形區,鋰離子難以在聚合物的分子長鏈上進行定向遷移,因此很難滿足室溫條件下的實際應用。PVDF和PVDF-HFP因具有相對寬的電化學窗口、良好的熱穩定性和較高的力學強度等優點,也是理想的聚合物電解質基體材料之一。Zhang等人報道了利用具有高離子電導率的PVDF基固態電解質實現金屬鋰枝晶抑制的相關工作。在所制備的電解質薄膜中,殘余的N,N-二甲基甲酰胺(DMF)溶劑并不是以自由分子的形式存在,而是與Li+發生配位形成了[Li(DMF)x]+的離子復合物。由于沒有自由溶劑分子的存在,PVDF基質會通過PVDF與DMF之間的相互作用來傳導[Li(DMF)x]+離子復合物,這種傳導機制與廣泛研究的PEO復合電解質十分類似。PVDF、鋰鹽和鍵合態的DMF分子之間存在著協同作用使得電解質的離子電導性能和全固態電池的電化學性能得到大幅改善。但是,對于DMF溶劑殘余含量的精確調控仍然難以把握。Xu等人采用了bottom-to-up的設計策略,利用四氫呋喃溶劑易于揮發的特質抑制了PVDF-HFP基聚合物電解質在制備過程中易發生的相分離的缺點,制備出了單一相的準離子液體/PVDF-HFP致密型聚合物電解質,該電解質具有1.55×10-3 S/cm的高離子電導率,3.4 MPa的拉伸強度,約1550%的斷裂伸長率。此外,作者還發現殘余的DMF溶劑并不是自由分子,而是與鋰鹽形成了[Li(DMF)3][TFSI]絡合物,有效提高了電解質的離子電導率與安全性。
除了以上這些被廣泛研究的聚合物基體,還有一些不同分子量的丙烯酸酯單體,如聚(乙二醇)甲基醚甲基丙烯酸酯(PEGMEA)和聚乙二醇丙烯酸(PEGDA),可以通過光固化或者熱固化的形式使用原位聚合的方法制備聚合物固體電解質,并且離子電導率可達10-4 S/cm級別。此外,脂肪族聚碳酸酯基的聚合物固態電解質由于其特殊的分子結構和高介電常數被認為是一類非常具有前途的聚合物電解質體系。Chai等人通過來自碳酸丙烯酯添加劑的正面界面效應為靈感制備了一種新型的聚碳酸乙烯基聚合物固體電解質,這種固體電解質具有良好的電化學穩定窗口且在50 ℃時離子電導率達到9.82×10-5 S/cm,用其裝配的高壓LiCoO2||Li電池具有良好的電化學性能和安全特性。聚碳酸丙烯酯(PPC)是一種由二氧化碳和環氧丙烷共聚反應得到的新型可降解脂肪族聚碳酸酯,每一個重復單元中都有一個極性很強的碳酸酯基團,具有低的玻璃化轉變溫度和高的熱尺寸穩定性等優點。Zhang等人受太極的啟發,提出了剛柔并濟的概念,用剛性的無紡布多孔膜負載柔性的聚合物離子傳輸材料PPC得到了復合固體電解質CPPC,其室溫離子電導率高達4.2×10-4 S/cm,這是由于PPC是無定形結構且分子鏈極易發生內旋轉,鋰離子主鏈上的羰基氧結合后鏈段運動較快從而導致鋰離子的快速遷移。
圖5是幾種不同電解質的平均性能的綜合分析,其中單一電解質系統總是顯示出一定的弱點,而復合電解質材料可以顯著改善性能。
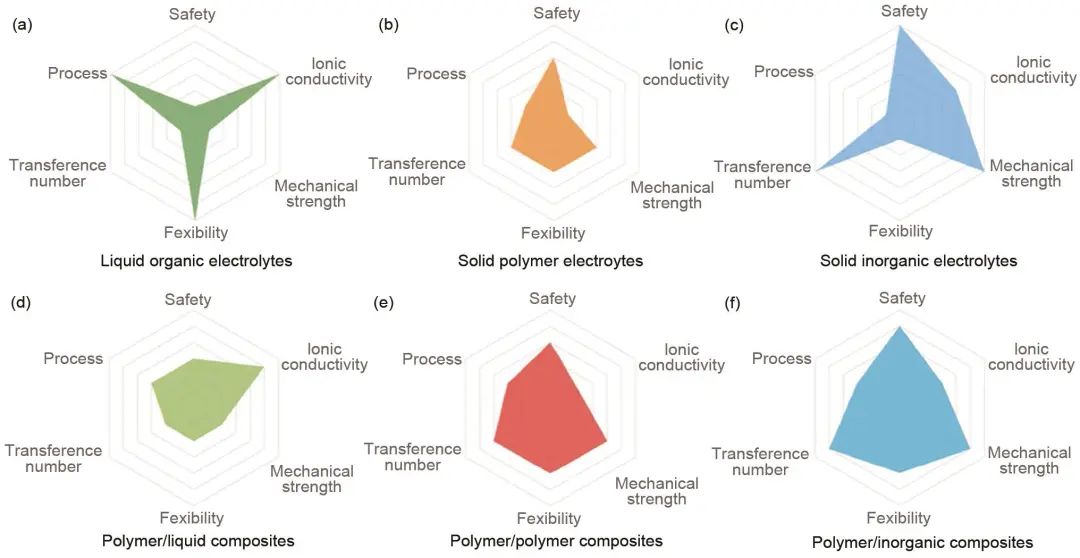
圖5 不同電解質的平均性能的雷達圖:(a) 有機電解質;(b) 固體聚合物電解質;(c) 固體無機電解質;(d) 聚合物/液體復合電解質材料;(e) 聚合物/聚合物復合材料以及(f) 聚合物/無機復合材料
3.2 復合聚合物固體電解質
目前常用的復合聚合物固體電解質制備策略主要包括:共混或交聯、加入支撐體、有機無機復合等。
3.2.1 共混或交聯
對聚合物基體進行共混或者交聯,通過降低聚合物的結晶度,可以有效提高復合聚合物固體電解質的室溫離子電導率。Lalia等采用共混制備了由四甘醇二甲醚、PEO、網狀纖維素和高氯酸鋰(LiClO4)組成的三元聚合物電解質。網狀纖維素和四甘醇二甲醚的加入有助于提高聚合物電解質的電導率和機械完整性,該電解質的室溫電導率最高可達10-4 S/cm。Khurana等報道了一種通過開環聚合用側鏈PEO交聯聚乙烯(PE)主鏈的固體電解質膜[圖6(a)],交聯抑制了PEO的結晶性,使電解質在室溫下具有高離子電導率(>1.0×10-4 S/cm)。另外,傳統的理論認為電解質膜需要具有足夠的機械強度之后才能有效抑制鋰枝晶的生長,但作者首次證明電解質膜抑制鋰枝晶生長的能力與其機械強度之間并不是絕對的關系。
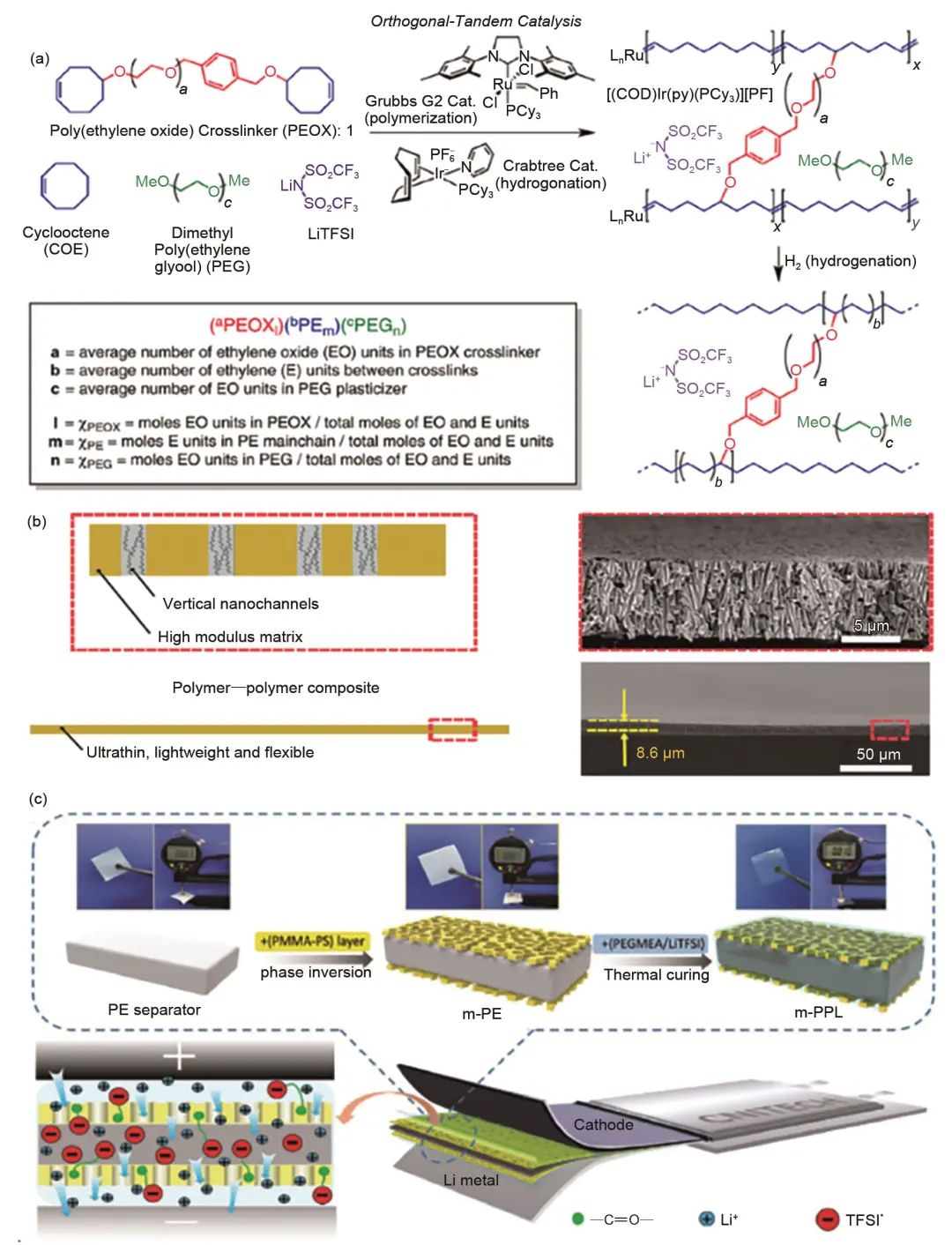
圖6 (a) 聚乙烯/聚環氧乙烷固體聚合物電解質的合成與命名;(b) 聚合物復合材料SSE的設計原理示意圖(左),超薄納米孔PI薄膜的橫斷面掃描電鏡圖像和排列整齊的納米孔的放大圖像(右);(c) m-PPL的制備工藝示意圖
3.2.2 加入支撐體
一般來說,單純的聚合物固體電解質在厚度和強度方面仍然差強人意,而加入薄而強勁的自支撐主體,可以實現較強的力學性能且厚度可控。Wan等報道了一種以多孔聚酰亞胺膜(PI)為主體、PEO/雙(三氟甲烷磺酰)鋰酰亞胺(LiTFSI)作為填充物的超薄、柔性、機械強度高的聚合物-聚合物SPE[圖6(b)],所制備電解質的厚度僅為8.6 μm。由于PI膜不易燃,機械強度高,可以有效防止電池在循環過程中由鋰枝晶引發的電池短路。經過物理刻蝕形成的垂直通道提高了注入的聚合物電解質的離子導電率(30 ℃下2.3×10-4 S/cm)。Wu等利用簡單的溶劑揮發法將PEO/LiTFSI聚合物電解質填充至聚乙烯隔膜的孔道內,制備了厚度僅為7.5 μm的超薄復合聚合物電解質。超薄的電解質縮短了鋰離子在電解質中的擴散時間和距離,并為電池提供了足夠的鋰離子。這些研究成果清楚地表明,通過引入強基體膜,聚合物固體電解質的電化學和力學性能得到了顯著提高。為了進一步改善電解質和電極之間的界面兼容性,Wang等報道了一種以改性PE為主體,聚乙二醇甲基醚丙烯酸酯(PEGMEA)和鋰鹽為填料的10 μm厚超薄聚合物固體電解質。多孔的聚甲基丙烯酸甲酯-聚苯乙烯(PMMA-PS)界面層緊密貼合在PE的兩側,有效地改善了電解質和電極之間的界面相容性,見圖6(c)。
3.2.3 有機-無機復合聚合物固體電解質
對于聚合物/無機物的復合電解質材料來說,早期的研究基本都圍繞在惰性填料上。Croce等將陶瓷納米顆粒(5.8~13 nm)首次引入到PEO基聚合物固體電解質。固體電解質的離子電導率提高了3個數量級,在30 ℃達到10-5 S/cm級別,這是得益于陶瓷納米顆粒大的表面積和路易斯酸的綜合效應的特點,可以防止PEO鏈在冷卻過程中(從高于60 ℃到環境溫度)再結晶。在聚合物電解質中摻雜惰性填料可以有效降低聚合物電解質的玻璃化轉變溫度,此外無機填料的加入可以有效改善電解質的力學性能。近十年來,研究者們的目光更多地轉向了活性填料,例如Li1+xAlxTi2-x(PO4)3(LATP),Li6.75La3Zr1.75Ta0.25O12(LLZTO)等。活性填料即參與導電過程的填料,由于其本身能夠提供導電的鋰離子,鋰離子不僅可以在聚合物相中轉移,還可以在活性填料相中轉移,這樣聚合物電解質的電導率將會有效提高。Huang等通過燒結制備出具有聯鎖多孔結構的Li0.33La0.557TiO3(LLTO)骨架材料,隨后將聚環氧乙烷(PEO)引入孔中,生成具有垂直雙連續相的PEO-LLTO框架固體電解質,離子電導率高達2.04×10-4 S/cm。該研究表明LLTO框架能夠通過內在空位快速傳輸Li+,同時低晶化的PEO表現出快速Li+轉移能力,協同實現高效Li+導電。Hu等發明了一種新穎的同步靜電紡絲/靜電噴霧方法來制備富有連續界面的薄膜超離子導體。由LLZTO和PAN所組成的超快鋰離子導體在25 ℃下可實現1.18×10-3 S/cm的離子電導率,超高離子電導率的關鍵在于構建了大量的連續界面,使快速界面傳導成為主導機制。這種新型的體界面超離子導體可使固態電池在室溫下實現低的極化以及長的循環壽命。Ju等通過原位聚合制備了聚碳酸酯(PVCA)與Li10SnP2S12(LSnPS)的復合固體電解質,聚合后得到了具有良好室溫電導率和寬電化學窗口的PVCA-LSnPS復合電解質。更重要的是復合電解質和鋰金屬負極以及固態正極之間都展現出良好的兼容性,極大地降低了電池的界面阻抗,從而獲得了優異的電化學性能。
4 硼氫化物固體電解質
4.1 硼氫化物固體電解質研究進展
金屬氫化物作為大規模儲氫的候選材料而為人們所熟知,它們通常由金屬陽離子(Li+,Na+,Mg2+)和絡合陰離子組成,具有低的晶界電阻、良好的還原穩定性、離子選擇性高、機械靈活性以及易于器件集成和低加工成本等特性,因此非常適合作為固態電解質應用于固態電池,但是由于其離子電導率不佳,組裝的固態電池性能方面一直未獲突破。早期的研究中,具有代表性的氫化物電解質主要包括:Li2NH、LiNH2、LiAlH4和Li3AlH6等,但是其離子電導率大多不超過1×10-4 S/cm,并且化學穩定性以及電化學穩定性差,其固態電池性能一直未獲突破,因此未受到廣泛關注。直到2007年,日本東北大學的Matsuo等發現硼氫化鋰(LiBH4)通過微波加熱可以實現從正交晶系到六方晶系的轉變,并且其離子電導率提高了3個數量級,達到了10-2 S/cm級別。然而,硼氫化鋰只有在高溫時才表現出高的離子電導率。為了提高硼氫化鋰的室溫離子電導率,以及其在全固態電池中的實用性,研究者們做了大量的工作,探索出了多種改性方式,如摻雜改性、有機-無機復合改性,以及水合、氧化處理等,并且開發出了一系列硼氫化鋰衍生物固態電解質。最近10年,尤其是最近5年,硼氫化物的離子電導率已達到了10-2 S/cm量級,甚至超過有機液態電解液的水平,更是激發了人們的研究興趣。
目前提高硼氫化物離子電導率或改善其化學以及電化學性質最常用的方法就是通過摻雜改性形成固溶體。Maekawa等發現,通過摻雜鹵化鋰(LiX,X = Cl,Br,I)可以大大降低其相變溫度,使其在低溫下仍具有高的離子電導率,并且,相比于Cl-和Br-,半徑較大的I―陰離子部分取代BH
后得到的LiBH4-LiI 固溶體具有最高的室溫離子電導率和最低的晶相轉變溫度[圖7(a)]。隨后,2013年,丹麥技術大學的Sveinbjo?rnsson等[98]仔細研究了溫度、成分對LiBH4-LiI 固溶體離子電導率的影響。他們的研究表明,當LiI的比例少于25%時,LiI的含量每增加10%,LiBH4-LiI固溶體的相變溫度就降低70 ℃;并且當LiI含量為40%時,其室溫離子電導率最高可達1×10-4 S/cm。2019年,都靈大學的Gulino等首次研究了三元體系內,鹵化物含量對LiBH4固溶體離子電導率和電化學穩定性的影響。當組分為Li(BH4)0.7(Br)0.2(Cl)0.1時,電導率最高可達1.3 ×10-5 S/cm(30 ℃),比純正交晶系的LiBH4的電導率高3個數量級。此外,他們的研究還表明鹵化除了提高離子電導率外,還能提高電解質的電化學穩定性,在僅含有Br-的體系[Li(BH4)0.7Br0.3]中,其電化學氧化窗口可達4.04 V(vs Li+ /Li)。除了形成固溶體改性之外,復合摻雜改性也是研究熱點,這種方法能夠綜合改善LiBH4固態電解質的離子電導率、電化學窗口和力學性能。Matsuo等通過結合兩種絡合陰離子BH4-與NH2-制備了Li2(BH4)(NH2)和Li4(BH4)(NH2)3,兩種硼氫化物的室溫離子電導率都達到了2 ×10-4 S/cm,如圖7(b)所示,比純的LiBH4提高了4個數量級。這是由于NH2-陰離子的引入為鋰離子引入了新的占據點位從而提高了鋰離子遷移率。
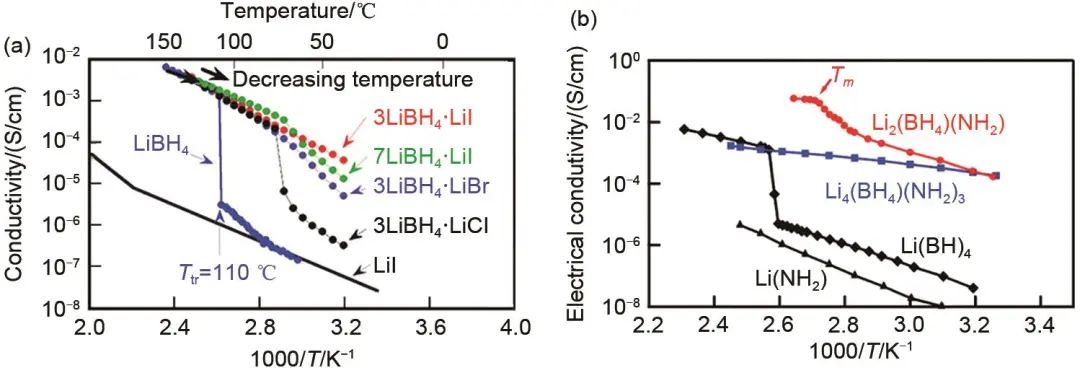
圖7 (a) LiBH4和LiBH4-LiX(X = Cl,Br,I)電導率和溫度的關系;(b) Li2(BH4)(NH2)和Li4(BH4)(NH2)3 離子電導率和溫度的關系
2015年,Shin-ichi Orimo課題組的Tang等首次制備出兩種碳硼烷型的固態電解質:LiCB11H12和NaCB11H12。在室溫下兩種電解質均為有序正交相,且與其類似物Li2B12H12和Na2B12H12相比,其向高溫相相變的溫度更低。電導率測試表明,403 K時LiCB11H12的離子電導率為0.15 S/cm,NaCB11H12在383 K時的離子電導率為0.12 S/cm。此外,直流電導率測量表明兩者陽離子遷移數接近于1。2016年,Tang等進一步發現LiCB9H10和NaCB9H10可以在室溫或者臨界室溫的溫度下保持高離子電導率的六方相,從而展現出媲美有機液體電解質的離子電導率(0.03 S/cm)。為了進一步提高離子電導率,Tang等通過復合CB9H
或CB11H
兩種籠狀陰離子從而形成陰離子合金鹽固溶體Li2(CB9H10)(CB11H12)和Na2(CB9H10)(CB11H12)。結果表明,這種陰離子合金化的固溶體可以在更寬的溫度范圍內抑制有序相的形成,從而有利于陽離子的快速傳輸。其中Na2(CB9H10)(CB11H12)展現了目前所報道的離子電導率的最高值,300 K時其離子電導率可達0.07 S/cm,即使在240 K,其離子電導率仍可保持在5×10-3 S/cm。這一工作的報道,極大地激發了人們對硼氫化物電解質的研究興趣。
4.2 硼氫化物在固態電池方面的應用
由于最近幾年硼氫化物在電導率以及電化學穩定性方面取得的進展,使其在固態電池的實用化方面展現了巨大的潛力,除了其令人印象深刻的離子電導率外,其作為固態電解質還有以下優勢:①輕質量。和氧化物、硫化物相比,由氫原子組成的硼氫化物具有最低的密度,這意味著采用硼氫化物作為電解質可以有效提升電池的功率和能量密度;②穩定的負極界面。硼氫化物和Li/Na/Mg金屬的相容性良好,可以確保穩定的電解質/負極界面;③高延展性。由于硼氫化物具有良好的延展性,通過簡單的冷壓工藝,電極和硼氫化物之間很容易形成致密的界面。
硼氫化鋰及其衍生物固態電解質具有一定的還原性,因此其與Li負極材料兼容性較好,但是其與氧化性的正極材料之間的問題比較突出,由于硼氫化物固體電解質電化學氧化物穩定性差,因此其與高壓正極之間往往會發生界面反應,形成不穩定的界面層。2013年,Takahashi等首次報道了以LiBH4為固態電解質的全固態鋰電池Li|LiBH4| LiCoO2。結果發現,固態電解質LiBH4與Li金屬負極能夠穩定兼容,然而與正極材料LiCoO2兼容性較差。還原性較強的固態電解質LiBH4會與正極材料LiCoO2發生氧化還原反應,導致界面穩定性變差。為了抑制硼氫化物電解質和氧化物正極之間的界面反應,Takahashi等接著采用脈沖激光沉積在電極材料LiCoO2和電解質LiBH4之間沉積了一系列的中間層(Li3PO4、LiNbO3以及Al2O3)。結果表明中間層的使用可以有效地抑制LiCoO2和LiBH4之間的界面反應,其中Li3PO4效果最好,使LiBH4和LiCoO2之間的界面電阻降低了3個數量級,LiBH4固體電解質電池的速率和循環性能都有了顯著的提高,這個結果表明通過引入人工界面層,可以使硼氫化物作為全固態電池的電解質在實際中得以應用。
基于硼氫化物固態電解質的全固態電池因其潛在的安全性和高性能電池等其他優點而迅速發展并引起越來越多的關注;然而,目前對硼氫化物基固態電池的基本原理,特別是從相關的界面行為、可用的電化學穩定性、電池成分和結構等方面的研究和認識還很有限。
5 鹵化物固體電解質
鹵化物固體電解質,因其高離子電導率和高電壓穩定性,引起了科學界的研究興趣。首先,一價鹵素陰離子與鋰離子的相互作用比二價硫或氧陰離子弱,因此有望實現鋰離子的快速傳輸。其次,鹵素陰離子的半徑較大導致在化合物中較長的離子鍵和更大的可極化性,進而有利于鋰離子的遷移和可塑性的提高。此外,離子性較強的無機鹵鹽在干燥空氣甚至在高溫下都能夠保持穩定。在有關鹵素無機電解質體系的前期報道中,高離子電導率與高穩定性往往不能兼得,這使得該體系所受關注相對較少。直到2018年日本松下公司首次報道了室溫離子電導率為10-3 S/cm的鹵化物固態電解質,隨后加拿大西安大略大學孫學良教授課題組首次實現在水溶液中合成高離子電導率的鹵化物固態電解質,鹵化物電解質再次引起研究者的廣泛關注。
在以往的研究中很少對鹵化物電解質進行系統的總結,其定義和分類也相對不明確,Li等根據鹵化物電解質LiaMXb中元素M的類別,將鹵化物電解質分為四類:第一類是M為第ⅢB族金屬(M=Sc,Y,La-Lu);第二類是M為ⅢA族金屬的鹵化物電解質(M=Al,Ga,In);第三類為其他二價金屬元素的鹵化物電解質(M= Ti,V,Cr,Mn,Fe,Co,Ni,Cu,Zn,Cd,Mg,Pb)。第四類是M為非金屬元素,如N、O、S等。不同類別鹵化物電解質的結構以及代表性鹵化物電解質的離子電導率如圖8所示。
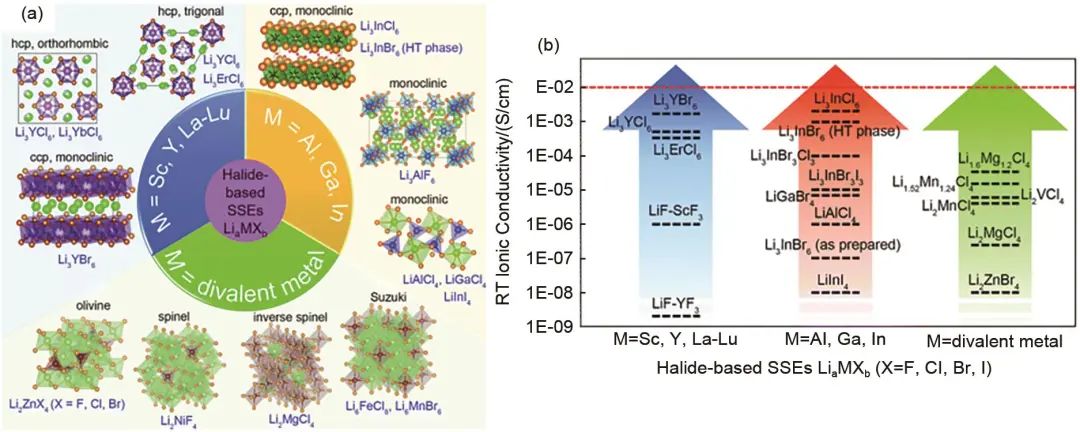
圖 8 (a) 含金屬元素鹵化物電解質分類;(b) 代表性鹵化物電解質室溫離子電導率
5.1 Ⅲ B族金屬鹵化物電解質
此類電解質的研究始于1984年,早期得到的電解質鋰離子電導率普遍偏低,未引起廣泛的關注。目前此類電解質的分子式分別為:LiMF4、Li3MCl6、Li3MBr6。理論計算預測一些該類電解質的室溫離子電導率可達10-2 S/cm。在已報道的含三價金屬的鹵化物電解質材料中,除了陰離子為氟離子,鹵素離子的半徑比陽離子半徑都要大。因此,結構中陰離子亞格子往往也是該晶體結構的結構框架。具有代表性的結構主要包括:陰離子六方密堆的正交相(hcp-O),陰離子六方密堆的三方相(hcp-T)以及陰離子立方密堆的單斜相(ccp-M)。此類材料中,最具代表性的是Li3MCl6和Li3MBr6。2018年,日本松下公司的Asano等首次報道了具有高室溫離子電導率的Li3YCl6和Li3YBr6,成功地將高離子電導率、高化學/電化學穩定性和可塑性結合在一起。室溫下Li3YCl6和Li3YBr6的離子電導率分別為0.51×10-3 S/cm和0.72×10-3 S/cm,熱處理后Li3YBr6的離子電導率可達1.70×10-3 S/cm,可以媲美石榴石型氧化物電解質。重要的是該體系實現了在接近實用的條件下應用于全固態電池中,選用目前商品化電池中成熟的未包覆鈷酸鋰作為正極材料,正極中活性物質的載量為10 mg/cm2,對應容量為1.5 mAh,所制備的固態電池首次庫侖效率可達94.8%,放電容量最高可達120 mAh/g并且在循環100次以內無明顯衰減。2020年,滑鐵盧大學Linda F. Nazara課題組中Park等報道了一種新的混合金屬鹵化物固態電解質Li3-xM1-xZrxCl6(M=Y、Er),其在25 ℃時具有高達2.04×10-3 S/cm的離子電導率。此外,這種氯化物電解質具有良好的電化學氧化穩定性,這使得可以使用未涂覆任何保護性涂層的>4 V級的正極材料而沒有任何明顯的氧化界面分解。組裝的全固態電池也展示了優良的電化學活性和循環性能,在3.0~4.3 V (vs Li/Li+)電壓范圍內,電流密度為0.55 mA/cm2 (0.5 C)進行恒流循環測試,在200次循環后,仍能保持高的容量和庫侖效率。
5.2 Ⅲ A族金屬鹵化物電解質
含ⅢA族元素(Al、Ga、In)鹵化物電解質的研究始于20世紀70年代,最早開發的是LiAlCl4,其離子電導率為1× 10-6 S/cm 。由于Al3+和Ga3+離子半徑較小,因此只能與大半徑的鹵素陰離子形成四配位的結構,如LiAlCl4、LiGaCl4、LiGaCl3、LiGab4、LiGaI4、LiGaI4等,而與小半徑的F-可以形成六配位結構,如:Li3AlF6、Li3GaF6等。而對于大半徑的In3+,與F-,Cl-,Br-均可形成六配位結構,如Li3InF6、Li3InCl6、Li3InBr6等,而與大半徑的I-只能形成四配位結構LiInI4。在幾十年的發展歷程中,由于Li-Al-X體系離子電導率過低,對其進行地研究較少,而含In體系(Li-In-X),尤其是Li-In-Br體系在早期研究得較多。
Li3InBr6及其衍生物發展多年,其室溫離子電導率最高可達1×10-3 S/cm,然而,該類材料熱穩定性差,在降溫過程中,高溫相會轉變成低溫相,并且其離子電導率也會降低到10-7 S/cm。目前,大部分的鹵化物電解質對水/濕度極其敏感。同時實現高離子電導率,對氧化物正極的高穩定性,水/濕度穩定性,甚至直接在水中合成是鹵化物固態電解質的重要目標。2019年,加拿大西安大略大學孫學良教授課題組中Li等報道了能夠直接在水中合成的Li3InCl6鹵化物固態電解質及全固態鋰電池中的應用。通過將LiCl和InCl3溶于水中初步干燥可得到Li3InCl6·2H2O中間體,進一步脫除結晶水即可得到目標產物Li3InCl6固態電解質(圖9),其室溫離子導為2.04×10-3 S/cm,并且在-25~75 ℃離子電導率測試范圍內沒有相轉變點。更重要的是,Li3InCl6固態電解質不僅具有較高的離子電導率,同時具有較好的水/濕度穩定性。機理分析表明,Li3InCl6和Li3InCl6·2H2O中間體在一定條件下的可逆轉換保證了其水/濕度穩定性。該策略合成簡便,可以在水溶液中批量合成。此外由于其良好的形變特性,可以用其包覆氧化物正極,以及作為空氣電池導電添加劑等。
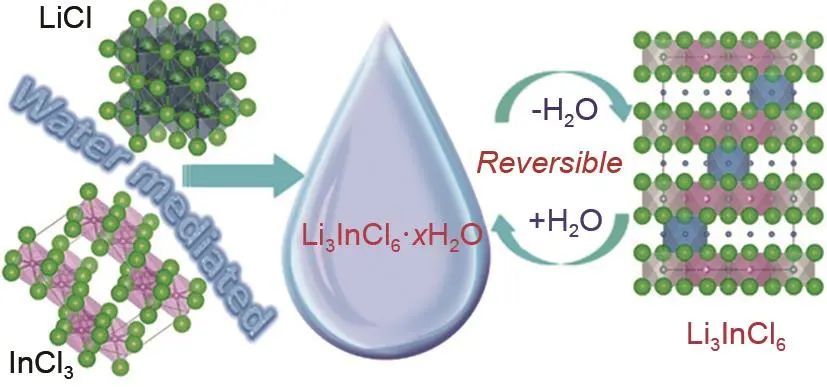
圖 9 水相合成Li3InCl6固態電解質示意圖以及Li3InCl6·xH2O與脫水的Li3InCl6之間的可逆轉化
5.3 二/四價金屬鹵化物電解質
二價金屬鹵化物電解質最早由Lutz及Kanno等報道,按結構主要分為四類:橄欖石結構,尖晶石結構,扭曲結構和Suzuki結構(Li6MX8)。此類材料室溫離子電導率普遍不高,目前最高僅能達到10-5 S/cm。一般而言,具有橄欖石和正尖晶石結構的鹵化物電解質其離子電導率低于具有反尖晶石結構的,反尖晶石結構的低于缺陷型反尖晶石結構的。另外根據其離子電導率變化趨勢,四面體位置的Li離子具有較高的流動性,部分的空位有利于降低Li離子移動的勢壘。二價金屬鹵化物電解質在一定程度上可以通過調節結構內部的空位、鋰離子濃度來提高其鋰離子電導率。和ⅢB族以及ⅢA族鹵化物電解質相比,這類鹵化物電解質的離子電導率仍然較低,進一步提高其離子電導率仍是目前研究的主要任務。
除了三價和二價陽離子之外,四價陽離子也被用于合成鹵化物電解質。Wang等用四價的Zr取代稀土元素制備了Li2ZrCl6固態電解質,克服了此類材料成本高昂和不耐潮氣的瓶頸。Li2ZrCl6和無包覆的單晶NMC811正極直接接觸所組成的全固態電池,在25 ℃、1C(200 mA/g)下循環200次之后,放電比容量仍達到了149 mAh/g,容量保持率達到98.6 %。
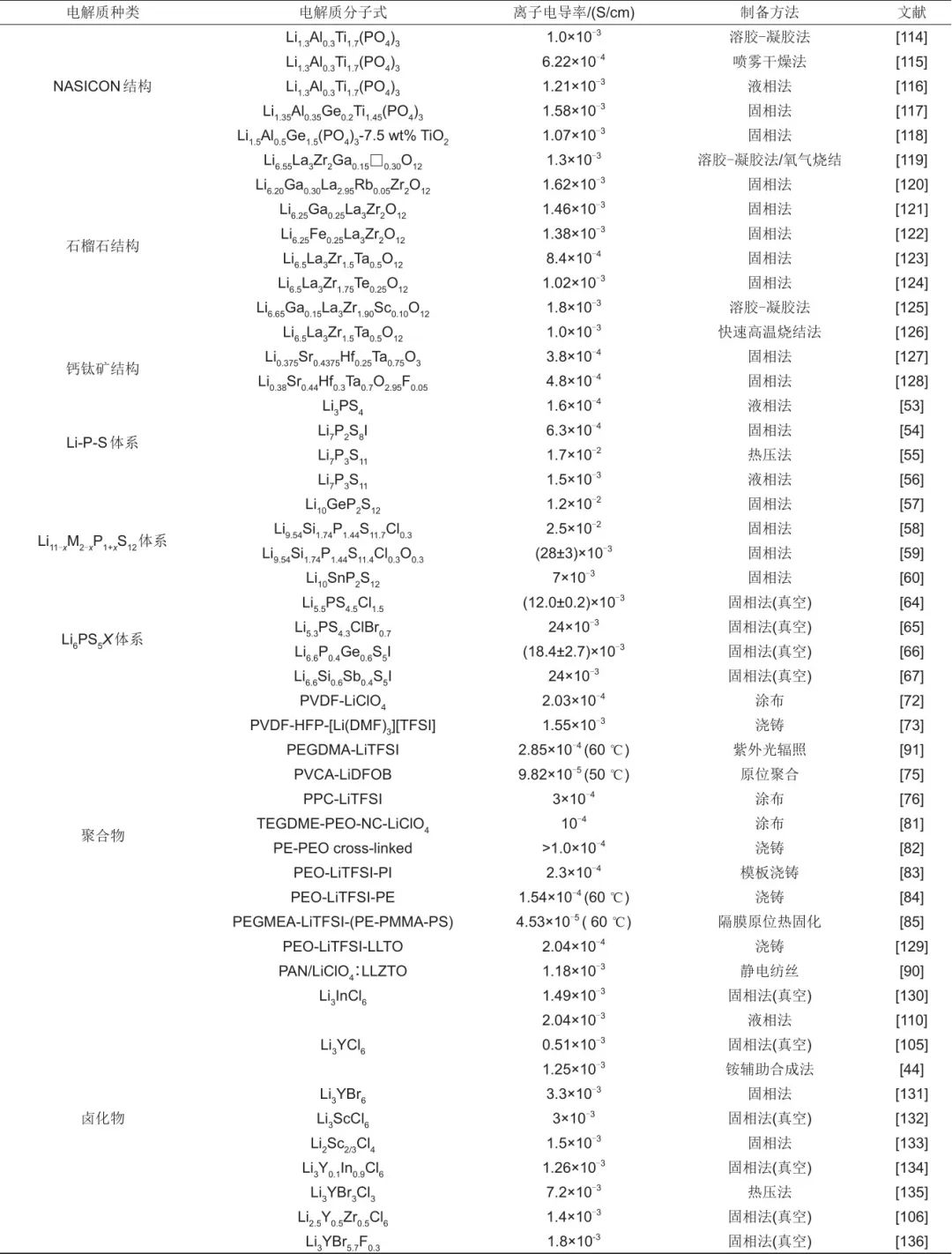
鹵化物電解質經歷了幾十年發展,由于其低的離子電導率一直未受重視,但是最近十年尤其是最近四年以來,多個材料的室溫離子電導率均實現了大于1×10-3 S/cm 的突破,再結合其良好的化學/電化學穩定性,以及其材料本身良好的可塑性,因此鹵化物電解質也是將來固態鋰電池實現大規模應用的一種潛在的候選材料。綜上,經過最近幾十年尤其是最近十年的發展,固體電解質的發展取得了巨大的進展,各類新穎的制備方法也不斷涌現,表3列舉了最近十年具有代表性的電解質的離子電導率以及制備方面的進展。
表3 不同類固體電解質的離子電導率以及制備方法
6 界面問題機理分析與改性策略
6.1 界面問題產生機理
界面穩定性是影響固態鋰電池電化學性能的重要因素之一,雖然最近幾年固體電解質的鋰離子電導率已經達到液態電解液的水平,但電池的容量仍然不高,循環和倍率性能遠低于傳統有機電解液電池,深入研究后發現決定電池容量和高倍率性能的關鍵因素除了固體電解質的離子電導率以及電解質的電化學窗口等電解質本身的原因之外,影響電池性能最主要的因素是電解質與正負極之間的界面問題。常見的界面問題主要包括空間電荷層、界面反應、界面接觸。
在正極側,由于固體電解質和電池正極間的熱力學不穩定性,兩者會通過發生界面反應形成界面層,并導致高的界面電阻。為了深入研究界面反應的可能原因,人們進行了許多探索。Haruyama等人通過理論計算研究了硫化物固體電解質/正極界面的陽離子的擴散特性。以LiCoO2/β-Li3PS4界面為例,計算結果表明鈷元素和磷元素交換能為負值,這說明界面處元素的擴散是自發的,擴散發生后會形成一個熱力學穩定的界面層。對于氧化物電解質,通常需要高溫燒結以保證固體電解質和正極之間的良好接觸,但是高溫下,固體電解質和正極之間會發生嚴重的化學反應和元素互擴散,導致生成難以控制的厚界面層,并且界面阻抗急劇升高。界面反應形成的界面層大多具有低的離子電導率,這將導致高的界面阻抗。此外,有的界面層還包含高電子電導成分,從而導致離子絕緣層的持續生長。因此,界面反應機理解釋了固體電解質和正極間產生高內阻并最終導致低倍率和循環性能的原因。此外,由于鋰電池活性物質通常是高離子電導率和電子電導率的混合體,而固體電解質是單一的離子導體。當正極材料和固體電解質接觸時,由于鋰離子在二者之間存在較大的化學勢差,鋰離子會從硫化物固體電解質向氧化物正極側遷移,形成空間電荷層。由于絕大多數正極材料在充放電過程中會伴隨著反復的體積變化,從而造成固體電解質和正極材料顆粒間的接觸缺失。界面接觸的缺失也會造成界面阻抗的增加和電池容量的損失。Koerver等人對比了充放電前后正極和硫化物固體電解質界面處的形貌發現,充放電前活性物質與電解質之間緊密接觸,經過50次充放電后,界面處出現明顯縫隙。固態電池中鋰金屬負極/電解質界面反應比較復雜,從熱力學角度出發,根據界面形成的特點固態電解質和鋰金屬負極的界面主要分為三種情況:①熱力學穩定的界面。②熱力學不穩定界面,一旦鋰金屬與固體電解質接觸,就會發生副反應,形成具有高離子電導和電子電導的混合導電相界面。混合導電界面層會和鋰金屬繼續反應,最終導致電池失效。③鋰金屬和固體電解質之間熱力學不穩定,但動力學上是穩定的,當界面產物電子電導率較低時,會形成亞穩態界面,這和傳統電池中SEI膜的形成過程類似。除了固體電解質和鋰負極的界面反應之外,近年來,鋰枝晶在固體電解質中的生長越來越引起了人們的重視。當電流密度和面容量過大時,鋰金屬會在固體電解質的內部和晶界處優先成核生長,并進一步滲透到其中,導致電池失效。
6.2 界面問題改性策略
針對固體電解質和各類電極材料間存在的各種問題,研究者提出了多種策略來改善界面穩定性,為了實現全固態電池的應用,引入界面修飾層被認為是最有效的方法。理想的界面修飾層應該有以下特點:高的離子電導率、低的離子電導率、寬的電化學窗口等,此外界面修飾層還應具有和正極接近的對鋰氧化還原電勢以及低的晶格失配度避免充放電過程中緩沖層脫離。常用的引入界面修飾層的手段包括:溶膠凝膠法、磁控濺射、原子層沉積(ALD)、激光脈沖沉積(PLD)等。常用的界面修飾材料包括LiNbO3,LiPO3,Li4Ti5O12,Al2O3等。此外高穩定性的電解質材料如氧化物,鹵化物也可以作為界面修飾材料。
硫化物電解質由于機械強度低、可塑性高,因此復合電極和固體電解質層都可以通過冷壓制備。但是由于硫化物和氧化物正極間多存在界面反應,因此通過對正極或者硫化物電解質進行界面改性可以有效降低界面阻抗。Li等采用原子層沉積技術將LiNbOx(LNO)包覆LiNi0.8Mn0.1Co0.1O2(NCM811)正極[(圖10(a)],其與Li10GeP2S12的界面穩定性得到了極大的提高。當LiNbOx緩沖層厚度為5 nm時,其電化學性能最好,與未包覆的LiNi0.8Mn0.1Co0.1O2的電極相比,包覆后,電極與電解質界面穩定,界面阻力小,循環穩定性明顯提高。Tatsumisago的團隊在這方面也做了大量工作來緩和正極和電解質之間的界面阻抗。以Li3PO4薄膜為例,通過室溫激光脈沖沉積的方法在LiNi0.5Mn1.5O4正極上沉積了一層Li3PO4薄膜[圖10(b)],組裝成固態電池后,未包覆的LiNi0.5Mn1.5O4無法實現可逆循環,而包覆后的循環良好,顯然,Li3PO4緩沖層明顯降低了正極和電解質之間的界面電阻。最近,一些新型的氧化物也被用于包覆高壓正極以提高其和硫化物電解質間的界面穩定性,Cao等采用簡單的濕化學方法,在LiNi1/3Mn1/3Co1/3O2和Li6PS5Cl之間的界面上包覆了一層15~20 nm的非晶態Li0.35La0.5Sr0.05TiO3。在非晶Li0.35La0.5Sr0.05TiO3的保護下,Li6PS5Cl的分解得到了有效抑制,因此大大降低了界面阻抗。Wang等利用LiCoO2和TiO2,LiCO3之間的氧化還原反應在LiCoO2表面原位形成了一層超薄的Li2CoTi3O8層。由于Li2CoTi3O8的引入而產生的Li2CoTi3O8/LGPS新界面與原始LiCoO2/LGPS界面相比具有更強的化學親和力,因此極大地降低了Li2CoTi3O8/LGPS的界面阻抗,有效提升了電池的循環穩定性。

圖10 (a) LNO包覆NCM811顆粒截面SEM圖譜;(b) Li3PO4包覆LiNi0.5Mn0.5O4顆粒SEM圖譜
對于氧化物電解質,為了避免混合正極燒結過程中電解質和正極間的界面反應,構建穩定混合導電界面,降低界面阻抗,應該發展自限性的界面形成方法,控制界面層的厚度以保證氧化物固體電解質和活性物質間電荷的輸運。Han等發現添加少量的B2O3作為助溶劑可以有效地降低LATP的燒結溫度(B2O3摻雜量為1%,生成產物縮寫LABTP),并有助于在LATP/LCO之間形成薄的、連續的混合導電界面(LABTP/LCO-LABTP)(圖11)。結果表明連續導電界面相的形成極大地改善了載流子的輸運性質,復合正極負載面容量高達6 mAh/cm。此外,這種方法同樣適用于高鎳正極,負載面容量高達10 mAh/cm2,并有望實現質量比能量大于400 Wh/kg的全固態電池。馬里蘭大學的Han等利用Li2CO3和Li2.3C0.7B0.3O3的反應在LiCoO2和LLZO電解質間形成了一層致密的Li2.3-xC0.7+xB0.3-xO3固體電解質層,極大地降低了復合正極的界面阻抗。與其他制備電解質層的方法不同,作者首先用Li2CO3分別包覆在LiCoO2和LLZO的表面,然后再高溫燒結,這樣避免了LiCoO2和LLZO直接接觸,從而避免了活性物質和電解質間的界面反應。采用這種全陶瓷復合正極的Li/LLZO/LCO全固態電池展現了高的循環穩定性和倍率性能。此外,大量的通過引入界面層的工作在最近幾年得以開展,使得固態電池電化學性能和循環性能有了大幅提升。相對于人們在抑制界面反應方面所做的研究,目前對固態電池體系中對正極材料本身的關注較少。Liu等通過原位電化學阻抗譜,聚焦離子束-掃描電子顯微鏡和固態核磁共振技術比較了傳統多晶NCM811(LiNi0.8Co0.1Mn0.1O2),小尺寸多晶NCM811和單晶NCM811材料在Li10SnP2S12硫化物電解質基的全固態電池中的長循環性能和電化學-力學行為。結果表明顆粒結構完整的單晶NCM811材料可提供連續的Li+擴散通道,使其在不同的上限電壓時均表現出高的比容量、穩定的充放電循環性能和出色的倍率性能。該工作通過系統的研究結果表明正極材料的結構完整性對全固態電池的循環穩定性和倍率性能起著不可忽視的作用。
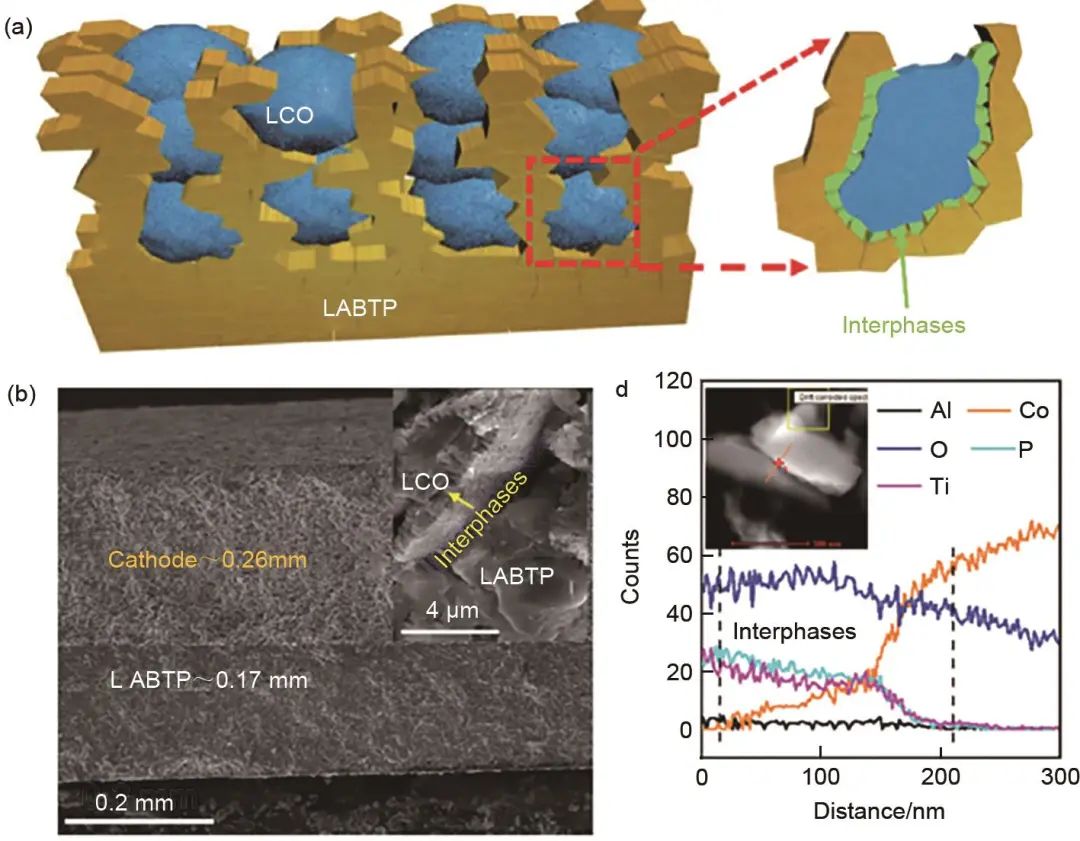
圖11 (a) 原位形成界面的共燒結復合正極示意圖;(b) LABTP/LCO-LABTP界面SEM圖譜和界面處元素線掃描圖譜
制備人工固體電解質膜也可以有效地抑制負極界面反應和枝晶生長。人工固體電解質膜可以避免高活性金屬鋰與固體電解質直接接觸,從而避免在界面發生不良的副反應。Zhang等通過H3PO4與鋰金屬的反應,在鋰金屬表面原位生成一層LiH2PO4保護膜,有效地提高了Li10GeP2S12的對鋰穩定性。Wang等在金屬鋰和硫化物電解質的界面處涂上一層塑料晶體電解質作為中間層,大大抑制了硫化電解質與金屬鋰的界面反應,此外塑料晶體電解質不可燃的特性還保證了電池的安全。電解質與正負極之間良好的浸潤性可以提升電池的界面穩定性,Yu等通過采用四亞甲基砜(TMS)選擇性地浸潤聚偏氟乙烯-聚醋酸乙烯基(PVDF-PVAC)復合電解質,TMS選擇性地與PVAC結合,在陰極和陽極界面上形成PVAC/TMS層,同時解決了鋰金屬電池中電解質與正負極界面接觸差的問題,極大地抑制了界面副反應以及鋰枝晶的生長問題。此外,PVAC/TMS界面層可以在陽極和陰極界面上構建有效的鋰離子傳輸路徑,提高了電池的倍率性能。為了抑制鋰枝晶生長并減小電池尺寸,三星電子的Lee等采用了一種沉積型鋰金屬負極技術,解決了影響電池壽命與安全性的枝晶問題,研究的核心思想是采用Ag-C納米復合層取代鋰金屬作為負極。結果表明在Ag-C納米復合層和不銹鋼(SUS)集流體之間可以形成致密的Li金屬層,作者認為Ag可與鋰合金化,從而降低鋰的形核勢壘,因此有助于將鋰均勻地沉積在集流體上。在充電過程的早期,Ag納米顆粒被鋰合金化,但有相當一部分Ag向集流體側移動,促進了鋰金屬的均勻沉積(圖12)。值得注意的是,作者還制備了的Ah級的軟包電池,體積能量密度可達900 Wh/L,并且可以穩定循環1000次。對未來發展高能量密度和高安全性固態電池具有啟發意義。
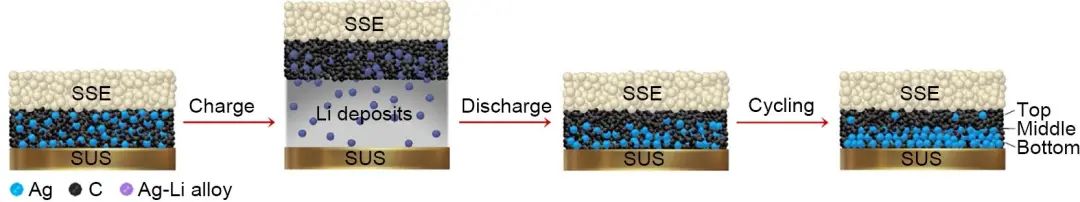
圖12 在充電和放電過程中,鋰金屬在用Ag-C納米復合層和集流體間沉積-剝離的過程示意圖
7 固態電池技術
目前兼具離子導電特性和力學特性的電解質制備技術尚不完備,并且電解質和電池正負極之間仍面臨嚴重的界面問題,為了提高固態電池能量密度以及早日實現全固態電池的實用化,各國的研究者嘗試了多種固態電池技術,涉及的研究重點主要包括:原位固態化、超薄電解質膜、正極表面包覆以及高面容量正極設計等。
考慮到現有液態鋰離子電池的安全性已經顯著提高,近期能夠規模化生產的有可能是介于液態鋰離子電池與全固態鋰電池之間的固/液混合型電池。該類電池可以首先在某些細分市場找到應用,隨著循環性能、安全性以及其他綜合技術指標的逐漸提升,逐步拓展到新能源汽車、規模儲能等大規模應用方面。在此基礎上,逐步減少液體或凝膠類電解質的比例。早在2012年,密歇根州立大學的Wang等就嘗試在LLZTO電解質中加入酯類電解液,結果表明液態電解質的加入極大地改善了LiFePO4|LLZTO|Li電池倍率性能以及循環穩定性。2018年,西安大略大學的Wang等系統研究了電解液用量對固態電池界面阻抗的影響,結果表明少量電解液的加入即可極大地降低LiFePO4正極和LATP電解質間的界面阻抗,并且相較于純度LATP電解質,固液電解質界面膜的形成可以有效地避免LATP被鋰金屬還原,并且鋰枝晶的生成得到有效抑制。此外,復旦大學的Wang等發現在固態鋰硫電池中加入少量電解液并不會誘導多硫化物穿梭或造成電池容量的衰減,相反,電解液加入后電池的循環壽命和穩定性有了極大地改善。Han等發現,通過原子層沉積的方法在石榴石電解質Li7La2.75Ca0.25Zr1.75Nb0.25O12 (LLCZN)表面沉積一層Al2O3可以極大地降低LLCZN與鋰金屬之間的界面阻抗。然而,全電池組裝過程中仍需加入少量的液態電解液[電解液的主要成分為1 mol/L LiPF6和FEC/FEMC/HFE (20∶60∶20,按體積計算)],以改善復合正極和石榴石電解質之間的界面浸潤性。
7.1 原位固態化
液態電解質的原位固態化技術也是一種提高電池性能非常實用的策略。Wang等開發了一種新型的凝膠聚合物電解質原位界面固化技術,解決了鋰-硫(硒)電池中電化學活性和穩定性之間的平衡問題,構筑出混合固液鋰-硫(硒)電池。作者將六氟磷酸鋰(LiPF6)引入到商品化的氧化鋁(Al2O3)/聚乙烯(PE)隔膜表面,從而得到了一種功能性的LiPF6/Al2O3/PE復合隔膜,并將此復合隔膜應用于準固態的鋰-硫(硒)電池。利用LiPF6原位引發電極/電解質界面上的陽離子聚合反應,使電池內部的醚類電解液前驅體發生梯度固化,在正極表面生成凝膠聚合物電解質層,同時在正極內部保留適量的液體電解質。這一原位固態化的策略既可以保證正極內部放電產物的適度溶解,提高正極材料的電化學反應活性,又能夠抑制可溶性中間體的溶出和“穿梭效應”,提高電池的循環穩定性,從而使鋰-硫(硒)電池同時具備高能量密度與長循環壽命優勢。Lu等提出了一種原位形成正極電解質界面膜(CEI)的方法,通過熱引發原位聚合碳酸乙烯(VC)以及同時引入CEI形成劑[二氟硼酸鋰(LiDFOB)]的方法,在陰極和固體電解質之間構建高壓穩定的界面層,使得PEO基固態鋰電池在高電壓下實現穩定循環。在3.0~4.2 V的電壓范圍內循環500次后,電池容量保持率為71.5% (101.6 mAh/g),而使用未修飾的LiCoO2電極的電池容量衰減迅速,循環200次后僅保持34.1 mAh/g的放電容量。此外,CEI修飾的LiCoO2/PEO-SPE/Li電池比大多數液態鋰離子電池和LiFePO4固體聚合物鋰電池具有更高的安全性,表明CEI涂層可以在很大程度上抑制高溫下正極與電解質之間的反應。Gao等利用含有多種鋰鹽的電解液在鋰金屬表明原位沉積了一層納米復合電解質界面膜,有效避免了鋰金屬和Li10GeP2S12電解質直接接觸,從而避免了循環過程中Li10GeP2S12的還原。由于該復合電解質是通過電解液在鋰表面原位分解形成的,因此具有良好的化學和電化學穩定性,對鋰和Li10GeP2S12均具有親和力,界面電阻較小,實現了超過3000 h的鋰對鋰循環,考慮到鋰鹽的多樣性,可以通過此方法制備多種復合界面膜來穩定固態電池界面。
7.2 超薄電解質膜
為了能在全電池水平上獲得比液態電解質鋰離子電池更高的能量密度,最直接的兩種方法是提高正極側活性物質的負載量或者降低固體電解質層的厚度。Wu等通過計算發現,在相同活性物質負載量(活性物質:NCA,厚度:60 μm,負載量19.5 mg/cm2)的前提下,為達到與鋰離子電池相同的質量能量密度,LLZO(5 g/cm3)、LATP(3 g/cm3)和LGPS(2 g/cm3)電解質的厚度需分別低于41 μm、74 μm、115 μm。目前,大部分無機固態電池主要是通過粉末冷壓的方法制備的,然而大多數電池只能通過降低正極活性物質的比例來獲得良好的電化學循環性能。為獲得合適厚度的薄層固體電解質,人們嘗試了不同的方法,主要包括脈沖激光沉積、溶膠-凝膠、氣溶膠沉積、射頻磁控濺射和原子層沉積等。LiPON由于具有較強的機械穩定性和致密性以及寬的電化學窗口[5.5 V(vs Li/Li+)],可有效抑制鋰枝晶的形成,并具有優良的循環穩定性,因此成為目前應用最成功的薄膜型固態電池用電解質。目前絕大多數LiPON薄膜的離子電導率在10-7~10-5之間。Lee等通過在氮氣下濺射(1-x)Li3PO4·xLi2SiO3靶材成功制備出LiSiPON薄膜電解質,通過提高Si的比例,離子電導率最高可達1.24 ×10-5 S/cm。除了磁控濺射方法以外,通過原子層沉積的方法可以方便地制備不同形狀的LiPON薄膜并可以提高其中的氮含量。此外,各種類型的高離子電導率固體電解質薄膜如NASICON、石榴石以及鈣鈦礦結構的固體電解質都可以通過物理和化學的方法成功制備。
采用固體電解質對隔膜進行改性可以有效抑制鋰枝晶的生長以及抑制鋰金屬與液態電解質間的副反應。Shi等將LATP涂敷聚丙烯隔膜(LATP-PP),并研究了復合改性隔膜的物理和電化學性質,結果表明LATP的引入可以有效提高隔膜的熱穩定性以及其和電解液間的潤濕性,此外由于包覆層中有更多鋰離子擴散通道。LATP-PP復合隔膜比Al2O3包覆PP膜具有更高的離子電導率。高離子電導率和界面穩定性使得LiNi0.5Mn1.5O4基5 V鋰離子電池具有良好的倍率容量和循環性能,表明LATP-PP復合隔膜在高壓電池中具有廣闊的應用前景。Yan等系統地研究了含鈦固體電解質(LLTO,LATP)改性隔膜對鋰電池電化學性能的影響。結果表明相較于Al2O3改性的隔膜,含鈦固體電解質改性隔膜更容易實現鋰金屬的均勻沉積,這是因為致密的固體電解質層可以有效抑制枝晶生長。此外,LATP@PE隔膜在Li|Cu電池中循環壽命最長,在NCM811|Cu電池中容量保持率最高。LATP和LLTO與鋰金屬之間反應的差異是造成阻抗差異的根本原因,LATP@PE在循環過程中形成了更穩定的SEI層,從而使其具有更好的電化學性能。因此,這些結果表明,用含鈦固體材料改性隔膜是一種有希望的金屬鋰電池優化策略。結合適當的電解質和適當的添加劑的使用,可以獲得更高性能的鋰金屬電池。
采用傳統工藝直接制備超薄電解質膜在實際應用中具有無可比擬的優勢,一方面可以通過電池的堆疊減少外包裝質量從而提高電池能量密度,另一方面可以利用傳統鋰離子電池現有的設備從而實現規模化生產。Jiang等采用傳統流延工藝結合高溫退火成功制備了厚度僅為25 μm的Li0.34La0.56TiO3 (LLTO)自支撐陶瓷膜[圖13(a)],相較于冷壓法制備的厚電解質片,LLTO薄膜的總離子電導率從9.6×10-6提升到2.0×10-5 S/cm,彎曲強度為264 MPa。當電解質膜厚度為41 μm時其彎曲強度即可滿足電池組裝要求,并且采用磷酸鐵鋰組裝的全固態電池初始放電容量為145 mAh/g,50次循環保持率為86.2%。在涂布工藝中,黏結劑對于活性物質的分散以及漿料成膜是必不可少的,但是在固態電池的制備過程中黏結劑一旦溶解,就會在活性物質或者電解質表明形成一層致密層,并起到電子和離子絕緣體的作用,從而增加電池內阻。對于氧化物基電解質可以通過高溫退火消除黏結劑的影響,但是對于硫化物基電解質,過高的溫度會引起電解質和黏結劑的反應,導致離子電導率下降,電子電導率升高。Yamamoto等發展了一種無黏結劑的片狀電池制備策略,其方法關鍵在于采用了揮發性的聚碳酸丙烯酯作為黏結劑用于分散硫化物電解質、活性物質和導電劑,并且成功獲得了正極,固體電解質和負極堆疊的三層薄片,然后通過低溫熱處理去除黏結劑。結果表明,黏結劑去除后,薄片型電池仍保持穩定的結構,并且電池的倍率性能和循環性能得到明顯的提升,電池能量密度比之前報道的片狀電池提高了2.6倍。但是在漿料涂布過程中,仍不可避免地使用了苯甲醚作為溶劑,這對硫化物電解質的穩定性有一定影響。
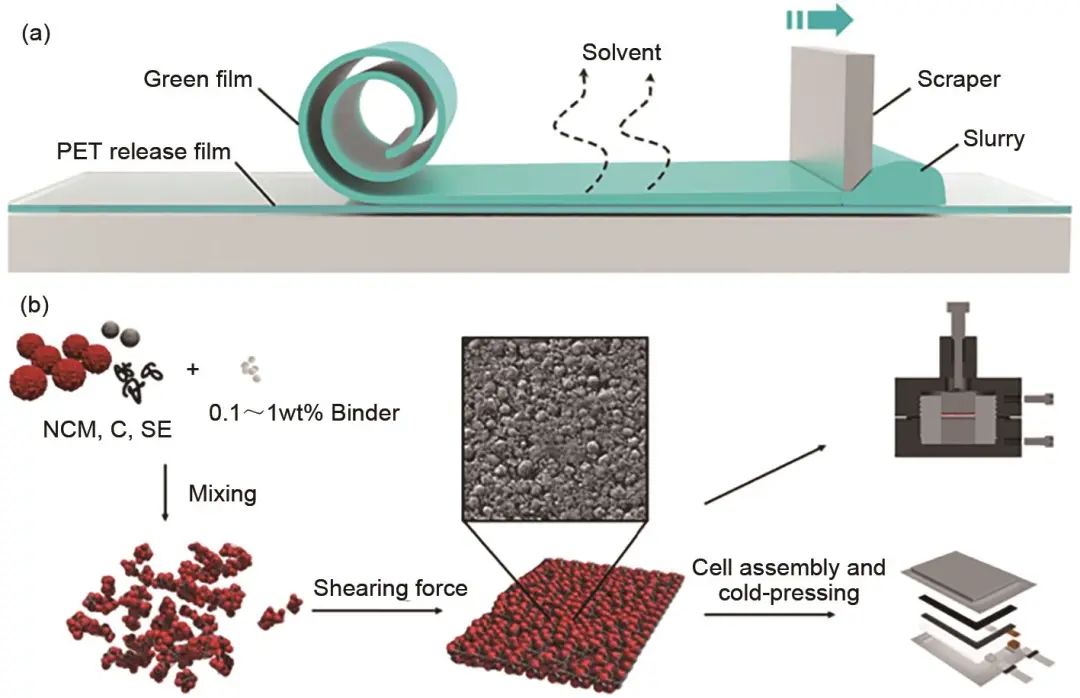
圖13 (a) 流延法制備LLTO膜示意圖;(b) 復合硫化物電解質/NCM正極膜示意圖
干法制備電極理論上比濕法制備的電極更有優勢,因為不使用溶劑可以避免黏結劑在活性物質或電解質表面形成致密膜,但是干法處理過程中黏結劑分子還是會通過增加孔隙率和阻塞離子/電子傳輸途徑來增加電池阻抗,因此,黏結劑的用量應盡量少。Hippauf等發展了一種通用的干法制膜的工藝,他們采用纖維狀的聚四氟乙烯作為黏結劑,由于聚四氟乙烯纖維狀的獨特結構可以有效地減少黏結劑對活性物質和電解質直接的表面堵塞,此外,其一維結構非常有助于成膜,即使黏結劑的用量降低到0.1%,仍可以獲得良好的獨立自支撐的電解質膜[圖13(b)]。因此采用這種方法極大地減少了非活性物質的使用,即使正極(LiNi0.9Co0.05Mn0.05O2)含量高達85%,所制備固態電池仍在室溫下展現了良好的倍率性能和循環穩定性。此外,這種方法具有良好的通用性,非常適合規模化生產以實現Ah級電池的制備,作者通過進一步優化電池設計制備了一個9 cm2的軟包電池,在沒有任何人工壓力的情況下,電池可以穩定地循環100次。Zhang等采用這種方法成功制備了超薄的Li5.4PS4.4Cl1.6硫化物電解質膜,厚度僅為30 μm,電導率高達8.4 mS/cm,并且具有良好的韌性,說明此方法具有良好的通用性。
7.3 正極表面包覆
開發高壓鋰離子電池是實現高能量密度鋰離子電池的關鍵,但是其過高的電壓會促使電解質分解。在正極表面包覆對高壓穩定的電解質涂層被證明是提高電池穩定性的一種非常有效的手段。Yang等采用簡單的液相法結合低溫固相反應,在LiCoO2表面包覆了一層厚度約為20 nm的致密LATP涂層,這種連續的致密膜可以防止LiCoO2與電解質直接接觸。與未包覆的LiCoO2相比,采用PEO基聚合物組裝固態鋰電池后,電池的循環和倍率性能有明顯改善,在4.2 V的充電截止電壓下,經過50次循環后,電池的放電容量保持率為93%,這說明連續致密的LATP電解質膜能有效抑制PEO在高電位下的氧化。Nie等利用微分電化學質譜結合密度泛函計算對LATP包覆LiCoO2的作用機制進行了深入分析。結果表明LiCoO2的表面催化作用是PEO基聚合物電解質在4.2 V下分解的根本原因。在LiCoO2表面包覆穩定的固體電解質LATP可以緩解這種表面催化效應,從而可以將穩定工作電壓提高到4.5 V。Wang等優化了LATP的包覆策略,進一步提升了包覆層的穩定性。作者先通過簡單的球磨法把LATP預包覆在LiCoO2表面,再進行退火處理,從而在LiCoO2表面原位形成高壓穩定的包覆層。深入研究發現,LiCoO2與LATP在高溫下反應,除了生成LiPO3外,還生成了尖晶石相的Co3O4、CoAlO3、Co2TiO4。尖晶石相的氧化物不但具有高壓穩定性,其晶格氧的氧化能力比層狀相的LiCoO2弱得多。此外,LiPO3是一種良好的鋰離子導體,在高壓下具有良好的化學穩定性。該方法有助于在LiCoO2表面構建具有良好離子傳導能力的均質高壓穩定層,并且可以廣泛用于其他高壓、高能量密度正極材料設計。
7.4 高面容量正極
為了提高活性物質的負載量,研究者們也嘗試了不同策略和方法。三維多孔陶瓷骨架因具有連續的鋰離子快速傳導網絡,且其自支撐結構能夠防止無機顆粒的團聚,近年來作為無機填料被廣泛地應用于復合電解質中。Fu等制備了一種三維雙層石榴石(Li7La3Zr2O12)固體電解質框架(圖14)。這種雙層固體電解質由高溫熱壓燒結的致密層和三維的多孔層組成,致密層厚度僅為30 μm,但仍保持良好的機械穩定性,從而使鋰金屬陽極能夠安全使用。較厚的多孔層不但可以容納更多的正極活性物質還為致密層提供了機械支撐。此外,致密層與多孔層之間的界面燒結良好,不但提供了完整的離子傳輸通道,其良好的機械完整性還可以適應正極充放電過程中的體積變化,如固體硫或者多硫化物正極。結果表明,硫正極的負載量可達7 mg/cm2,并且準固態的鋰電池展現了較高的庫侖效率和循環穩定性。這種復合固體電解質的設計不但可以有效緩解硫正極的體積變化,致密層還可以有效緩解多硫化物的穿梭。雖然這種方法面向商用還需要進一步簡化制備工藝,但是這種多孔正極/致密電解質層的一體化設計思路為我們實現高面比容量全固態鋰電池提供了有益指導。類似地,Zhang等用LAGP電解質設計了一種新的三層三維多孔固體電解質,其中三維的LAGP層作為LiNi0.8Mn0.1Co0.1O2 (NCM811)正極的宿主,致密的LAGP薄層用于離子傳導可抑制枝晶生長,此外還在LAGP和鋰金屬界面處引入了一層聚合物電解質以改善和鋰負極的接觸問題。在這種結構下,多孔的LAGP層的正極負載量可達13.1 mg/cm2,0.1 C電流下可提供2.01 mAh/cm2的可逆面容量,循環50次的保持率約為70.0%,而傳統的二維電極只能提供0.11 mAh/cm2的低面積容量,且容量快速衰減。
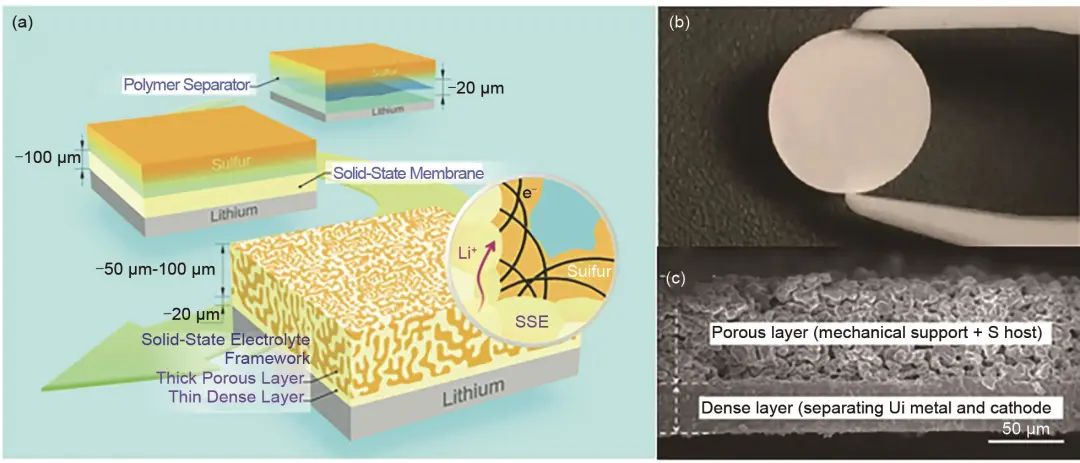
圖14 (a) 新型雙層固體電解質框架與傳統的聚合物隔膜以及剛性固體電解質對比示意圖;(b) 雙層固體電解質燒結后照片;(c) 雙層固體電解質燒結后界面SEM圖譜
除了多孔的電解質層的設計思路之外,在正極層和電解質層引入支撐體可以有效提高活性物質的負載量并改善電解質層的力學性能。Xu等分別采用不銹鋼網和Kevlar無紡布分別作為正極和電解質的支撐體獲得了一種具有高能量密度的全固態鋰硫電池。其中不銹鋼網負載的Li2S正極,負載量可達7.64 mg/cm2,而Kevlar無紡布支撐的Li3PS4 (LPS)電解質厚度僅為100 μm。因此,獲得的全固態鋰硫電池在室溫下展現了高的放電容量,良好的倍率性能以及循環性能,并且首次循環比能量高達370.6 Wh/kg(不包含集流體)。
7.5 固態電池產業化進展
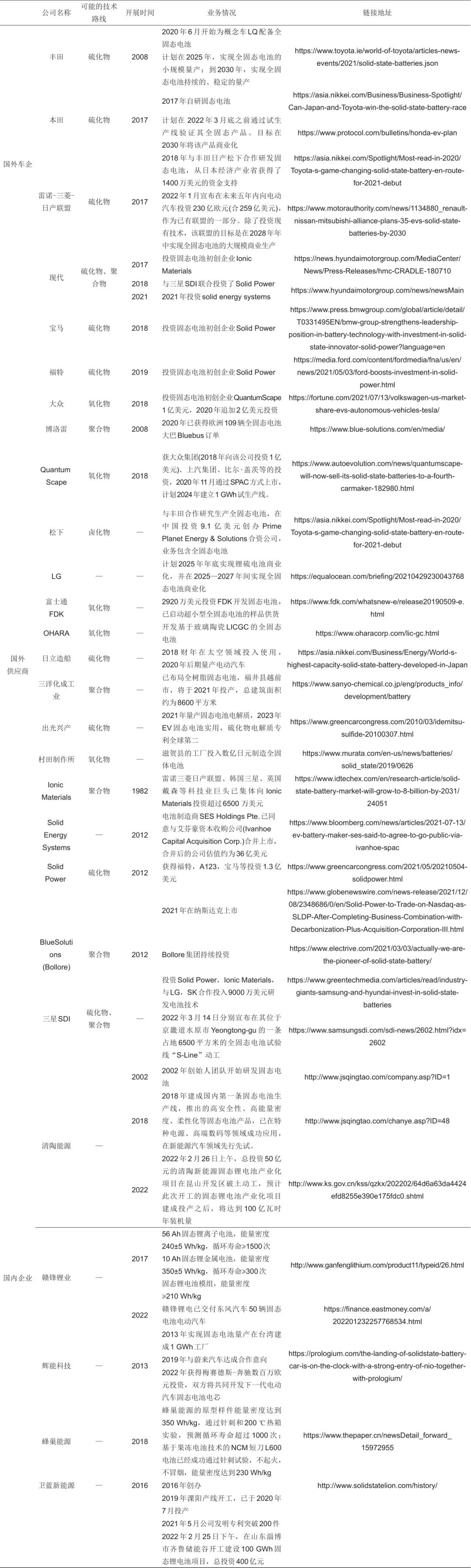
綜上所述,全固態電池可以從根本上解決現有鋰電池的安全問題,但是全固態電池實現產業化還有很長的路要走。目前,雖然一些其他類型的電解質也取得了不錯的進展,但是就技術成熟度而言,目前主流的技術路線仍然以聚合物、氧化物和硫化物為主,這三種材料體系都是20世紀80年代就開始的研究,因此經過幾十年的積累,各種技術路線上的代表性材料都已經篩選出來了。從固態電池全球的發展來看,目前固態電池的研發主要集中在中日韓美歐五個國家和地區,從發展趨勢來看采取從固液混合半固態電池到全固態電池過渡的形式分步實現最為可行。基于固態鋰電池發展的良好前景,除了傳統的新能源企業以及車企紛紛布局固態鋰電以外,眾多初創公司也雨后春筍般地成立,因此,我們整理了國內外企業在固態鋰電池產業化方面的布局情況,具體如表4所示。
表4 國內外企業固態電池產業化進展
8 結論與展望
總體來說,固態鋰電池是電池科研與工業界公認的下一步電池發展的主流方向,但是具體到固體電解質的電導率、化學/電化學穩定性、電極與電解質間的界面問題等科學問題,要實現產業化所面臨的規模化制備、成本控制等問題,以及要實現固態以及全固態鋰電池的真正應用,仍面臨眾多挑戰。在今后的研究工作中仍有下述問題值得注意。
(1)電解質
全固態鋰電池的發展主要依賴于固體電解質的發展,在經歷了緩慢的發展期后,在最近十年迎來了快速發展的黃金時間。目前最具潛力的電解質有氧化物、硫化物和聚合物,硼氫化物和鹵化物在最近五年也都有突破性進展。但是目前各類電解質的優缺點都很明顯,例如目前只有部分硫化物電解質的離子電導率接近或者超過有機液體電解液的水平,但因界面等方面的問題,其電化學穩定性尚不盡如人意;氧化物電解質雖具有良好的電化學穩定性,但鑒于其本身高的機械強度,通常需要高溫燒結來確保良好的界面接觸。沒有一種完美的電解質可以滿足應用所需要求,因此還需要改善各類電解質以獲得良好的綜合性能,此外還需要繼續努力探索發現新結構的離子導體,使用高通量篩選計算結合實驗將有助于發現新的化合物。
(2)表征手段
為了實現高安全、高比能量全固態電池的應用,還需要發展一系列與固體電解質相適應的表征手段,諸如用于觀察細微結構變化的冷凍電鏡、電池超聲掃描,固態核磁共振技術,以及準彈性中子衍射技術,原位技術(原位X射線粉末衍射儀、原位掃描電子顯微鏡、原位透射電子顯微鏡、原位原子力顯微鏡)等。這些將有助于加深對離子傳導機制、鋰枝晶生長機理、結構相變原理、界面微結構動態演變等的理解。
(3)界面
全固態電池中電極/電解質固固界面問題仍是目前全固態電池實用化過程中面臨的最實際的問題。包括界面阻抗大、界面穩定性不良、界面應力變化等,直接影響電池的性能。因此,需要設計具有優良鋰離子傳輸能力的界面,新的界面不但需要具有高的離子電導率還需要和正極以及電解質都具有良好的相容性。常用的手段包括表面包覆、調控復合正極組分等。此外還需要探索更適合固態電池的電極材料和表面包覆材料,同時需要對晶界和電極/電解質界面反應和動力學進行更深入的理解。(4)鋰金屬負極
與傳統鋰離子電池相比,只有采用鋰金屬負極的固態電池在全電池水平上的能量密度才更具競爭優勢,但是目前研究表明固體電解質的高剪切模量不能完全有效地抑制鋰枝晶,鋰金屬會沿著顆粒間的晶界生長,高反應活性電解質和鋰負極間的界面反應形成的混合離子電導相會極大地增大界面阻抗,從而造成電池失效。因此,很難通過一種方法解決枝晶問題。鑒于此,構筑具有高的界面能、低的電子電導以及具有一定自修復功能的人工界面層對實現鋰金屬負極的應用具有重要意義。
(5)全電池結構設計
一個有效的電池系統需要正極、電解質、負極等各部分之間的兼容。對于不同類型電解質,由于電極和電解質材料的特殊理化性質,通過將活性材料、固體電解質和導電碳混合在一起,很容易得到復合陰極層。而簡單的混合很難實現循環過程中穩定性高的互連電子/離子網絡。此外,固態電池中離子的傳輸強烈依賴于固體顆粒的致密接觸。而這些點接觸對電化學循環過程中產生的應力非常敏感,應力會導致裂縫的產生,引起界面接觸不良。為了避免復合材料內部空隙的產生,需要提高復合材料的致密度,這就需要優化復合材料的組成以及新的混合策略和成型技術。因此,探索新的技術,特別是結合現有的制造技術和工藝,實現電池結構的整體設計勢在必行,這對促進全固態電池的實際應用具有重要作用。
審核編輯:湯梓紅










 工商網監
工商網監
評論