


基于邊合成邊測序(Sequencing By Synthesis,SBS)技術(shù),Illumina HiSeq2500高通量測序平臺(tái)對(duì)cDNA文庫進(jìn)行測序,能夠產(chǎn)出大量的高質(zhì)量Reads,測序平臺(tái)產(chǎn)出的這些Reads或堿基稱為原始數(shù)據(jù)(Raw Data),其大部分堿基質(zhì)量打分能達(dá)到或超過Q30。Raw Data通常以FASTQ格式提供,每個(gè)測序樣品的Raw Data包括兩個(gè)FASTQ文件,分別包含所有cDNA片段兩端測定的Reads。
FASTQ格式文件示意圖如下:

FASTQ格式文件示意圖
注:FASTQ文件中通常每4行對(duì)應(yīng)一個(gè)序列單元:第一行以@開頭,后面接著序列標(biāo)識(shí)(ID)以及其它可選的描述信息;第二行為堿基序列,即Reads;第三行以“+”開頭,后面接著可選的描述信息;第四行為Reads每個(gè)堿基對(duì)應(yīng)的質(zhì)量打分編碼,長度必須和Reads的序列長度相同。
測序堿基質(zhì)量值
堿基質(zhì)量值(Quality Score或Q-score)是堿基識(shí)別(Base Calling)出錯(cuò)的概率的整數(shù)映射。通常使用的Phred堿基質(zhì)量值公式為: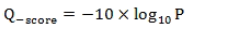
公式中,P為堿基識(shí)別出錯(cuò)的概率。下表給出了堿基質(zhì)量值與堿基識(shí)別出錯(cuò)的概率的對(duì)應(yīng)關(guān)系:
表1 堿基質(zhì)量值與堿基識(shí)別出錯(cuò)的概率的對(duì)應(yīng)關(guān)系表

堿基質(zhì)量值越高表明堿基識(shí)別越可靠,堿基測錯(cuò)的可能性越小。比如,對(duì)于堿基質(zhì)量值為Q20的堿基識(shí)別,100個(gè)堿基中有1個(gè)會(huì)識(shí)別出錯(cuò);對(duì)于堿基質(zhì)量值為Q30的堿基識(shí)別,1,000個(gè)堿基中有1個(gè)會(huì)識(shí)別出錯(cuò);Q40表示10,000個(gè)堿基中才有1個(gè)會(huì)識(shí)別出錯(cuò)。
以測序循環(huán)為單位,對(duì)單個(gè)樣品所有Reads平行測序的堿基質(zhì)量值做分布圖,可以查看單個(gè)樣品各個(gè)測序循環(huán)及整體的測序質(zhì)量。
堿基質(zhì)量值分布圖
注:橫坐標(biāo)為測序堿基在Reads上的位置,縱坐標(biāo)為堿基質(zhì)量值。顏色深淺表示堿基比重,顏色越深,說明該位置測定的堿基中為對(duì)應(yīng)質(zhì)量值的堿基所占的比重越大,反之亦然。
測序質(zhì)量控制
FASTQ文件中測序Reads需要與指定的參考基因組進(jìn)行序列比對(duì),定位cDNA片段在基因組或基因上的位置。在序列比對(duì)之前,首先需要確保這些Reads有足夠高的質(zhì)量,以保證后續(xù)分析的準(zhǔn)確。測序質(zhì)量控制方式如下:
(1) 去除測序接頭以及引物序列;
(2) 過濾低質(zhì)量值數(shù)據(jù),確保數(shù)據(jù)質(zhì)量。
經(jīng)過上述一系列的質(zhì)量控制之后得到的高質(zhì)量Reads或堿基,稱為Clean Data。Clean Data同樣以FASTQ格式提供。
測序數(shù)據(jù)產(chǎn)出統(tǒng)計(jì)
某項(xiàng)目各樣品數(shù)據(jù)產(chǎn)出統(tǒng)計(jì)見下表:
表2 樣品測序數(shù)據(jù)評(píng)估統(tǒng)計(jì)表
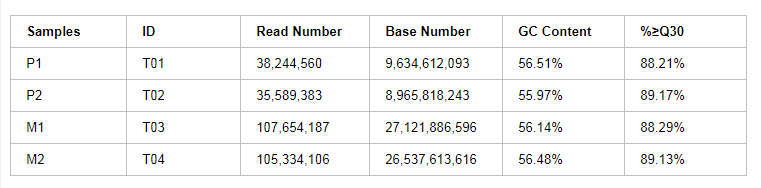
注:Samples:樣品信息單樣品名稱;ID:樣品編號(hào);Read Number:Clean Data中pair-end Reads(雙末端測序)總數(shù);Base Number:Clean Data總堿基數(shù);GC Content:Clean Data GC含量,即Clean Data中G和C兩種堿基占總堿基的百分比;%≥Q30:Clean Data質(zhì)量值大于或等于30的堿基所占的百分比。
轉(zhuǎn)錄組數(shù)據(jù)與參考基因組序列比對(duì)
獲得Clean Reads后,將其與參考基因組進(jìn)行序列比對(duì),獲取在參考基因組或基因上的位置信息,以及測序樣品特有的序列特征信息。
TopHat2是一個(gè)高效的序列比對(duì)軟件。它以高通量Reads比對(duì)軟件Bowtie為基礎(chǔ),將轉(zhuǎn)錄組測序Reads比對(duì)到基因組上,然后通過分析比對(duì)結(jié)果識(shí)別外顯子之間的剪接點(diǎn)(Splicing Junction)。這不僅為可變剪接分析提供了數(shù)據(jù)基礎(chǔ),還能夠使更多的Reads比對(duì)到參考基因組,提高了測序數(shù)據(jù)的利用率。
轉(zhuǎn)錄組測序數(shù)據(jù)中,只有比對(duì)到參考基因組上的數(shù)據(jù)才能用于后續(xù)分析。因此,將比對(duì)到指定的參考基因組上的Reads稱為Mapped Reads,對(duì)應(yīng)的數(shù)據(jù)稱為Mapped Data。
比對(duì)效率統(tǒng)計(jì)
比對(duì)效率指Mapped Reads占Clean Reads的百分比,是轉(zhuǎn)錄組數(shù)據(jù)利用率的最直接體現(xiàn)。比對(duì)效率除了受數(shù)據(jù)測序質(zhì)量影響外,還與指定的參考基因組組裝的優(yōu)劣、參考基因組與測序樣品的生物學(xué)分類關(guān)系遠(yuǎn)近(亞種)有關(guān)。因此,通過比對(duì)效率,可以評(píng)估所選參考基因組組裝是否能滿足信息分析的需求,及后期數(shù)據(jù)分析的可靠性。
各樣品測序數(shù)據(jù)與所選參考基因組的序列比對(duì)結(jié)果統(tǒng)計(jì)見下表:
表3 Clean Data與參考基因組比對(duì)結(jié)果統(tǒng)計(jì)表
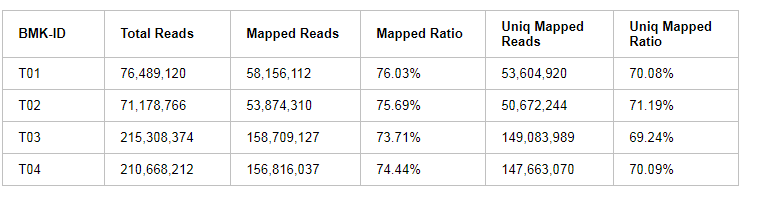
注:ID:樣品編號(hào);Total Reads:Clean Reads數(shù)目,按單端計(jì);Mapped Reads:比對(duì)到參考基因組上的Reads數(shù)目;Mapped Ratio:比對(duì)到參考基因組上的Reads在Clean Reads中占的百分比;Uniq Mapped Reads:比對(duì)到參考基因組唯一位置的Reads數(shù)目;Uniq Mapped Ratio:比對(duì)到參考基因組唯一位置的Reads在Clean Reads中占的百分比。
比對(duì)結(jié)果作圖
將比對(duì)到不同染色體上Reads進(jìn)行位置分布統(tǒng)計(jì),繪制Mapped Reads在所選參考基因組上的覆蓋深度分布圖。
樣品T01的Mapped Reads在參考基因組部分染色體上的覆蓋深度分布圖如下:
Mapped Reads在參考基因組上的位置及覆蓋深度分布圖
注:橫坐標(biāo)為染色體位置;縱坐標(biāo)為覆蓋深度以2為底的對(duì)數(shù)值,以10kb作為區(qū)間單位長度,劃分染色體成多個(gè)小窗口(Window),統(tǒng)計(jì)落在各個(gè)窗口內(nèi)的Mapped Reads作為其覆蓋深度。
理論上,來自成熟mRNA的Reads應(yīng)該比對(duì)到外顯子區(qū)。但是,由于以下原因一部分Reads會(huì)比對(duì)到內(nèi)含子區(qū)和基因間區(qū):
(1) 樣品提取時(shí)將含有Ploy(A)尾而內(nèi)含子沒有切除完全的mRNA(即mRNA前體)提出,使得來自內(nèi)含子片段的Reads比對(duì)到了內(nèi)含子區(qū);
(2) 基因組注釋錯(cuò)誤,原來為外顯子的區(qū)域注釋成了內(nèi)含子區(qū),或者相反;
(3) 基因組注釋水平低,對(duì)于使用轉(zhuǎn)錄組測序數(shù)據(jù)進(jìn)行的基因組注釋,由于轉(zhuǎn)錄組測序不能遍歷所有的時(shí)間和空間點(diǎn),使得用于注釋的轉(zhuǎn)錄組測序數(shù)據(jù)中不表達(dá)或低表達(dá)的基因剛好在該項(xiàng)目的樣品中檢測到較高豐度時(shí),來自這類基因的Reads就比對(duì)到了被注釋的基因間區(qū),這也是新基因和新轉(zhuǎn)錄本發(fā)掘的基礎(chǔ)之一;
(4) 測序樣品與參考基因組存在差異,比如測序樣品中突變形成新的轉(zhuǎn)錄組起始位點(diǎn)形成樣品特有的新基因,或者剪接位點(diǎn)差異形成新的轉(zhuǎn)錄本,這也是新轉(zhuǎn)錄本發(fā)掘的基礎(chǔ)之一。
統(tǒng)計(jì)Mapped Reads在指定的參考基因組不同區(qū)域(外顯子、內(nèi)含子和基因間區(qū))的數(shù)目,繪制基因組不同區(qū)域上各樣品Mapped Reads的分布直方圖,如下:
基因組不同區(qū)域Reads分布直方圖
注:圖中每個(gè)直方柱表示一個(gè)樣品,粉色區(qū)域?yàn)橥怙@子區(qū)、綠色區(qū)域?yàn)榛蜷g區(qū)、藍(lán)色區(qū)域?yàn)閮?nèi)含子區(qū),區(qū)域的高度表示比對(duì)到該區(qū)域的Mapped Reads在所有Mapped Reads中所占的百分比。
編輯:hfy
-
sbs
+關(guān)注
關(guān)注
0文章
15瀏覽量
12664 -
質(zhì)量控制
+關(guān)注
關(guān)注
0文章
28瀏覽量
8576
發(fā)布評(píng)論請(qǐng)先 登錄
原邊反饋AC/DC控制芯片中的關(guān)鍵技術(shù)
電源設(shè)計(jì)中的原邊反饋控制和副邊反饋控制方案分析
什么是高低邊開關(guān),高低邊開關(guān)怎么設(shè)計(jì)?
輪邊驅(qū)動(dòng)電機(jī)專利技術(shù)發(fā)展
SMT車間管理與質(zhì)量控制技術(shù)(續(xù)完
基于AC/DC控制芯片的原邊反饋技術(shù)
手機(jī)如何實(shí)現(xiàn)一邊充電一邊聽歌(邊充邊聽)呢

基于AC/DC控制芯片的原邊反饋技術(shù)
基于AC/DC控制芯片的原邊反饋技術(shù)






 工商網(wǎng)監(jiān)
工商網(wǎng)監(jiān)

評(píng)論