 一種量子化學與分子力學結合的方法
一種量子化學與分子力學結合的方法
本專題將介紹一種量子化學與分子力學結合的方法(QM/MM方法),該方法既包括量子化學的精確性,又利用分子力學的高效性,其基本思想是用量子力學處理感興趣的區域,其余部分用經典分子力學來處理。本章節以一個典型的沒食子酸分子(Gallic Acid,GA)為例,介紹輸入文件準備,QM/MM單點計算,QM/MM結構優化,QM/MM激發態計算。其中,BDF程序主要完成量子化學計算部分,其余部分由BDF開發成員修改的pDynamo2.0程序包完成。同時介紹如何讀取數據用于結果分析,幫助用戶深入了解BDF軟件的使用。
輸入文件準備
一般來說,QM/MM計算之前,需要對目標體系進行分子動力學模擬,得到適合的初始構象。當采用PDB、MOL2或xyz文件作為輸入時,pDynamo2.0程序包僅支持OPLS力場,對于小分子和非標準氨基酸力場參數不全,不推薦使用。建議優先采用Amber程序,通過拓撲文件輸入力場參數。以Amber為例,從動力學模擬軌跡提取感興趣的結構存儲于.crd文件中,與對應的參數/拓撲文件.prmtop一起可以作為QM/MM計算的起始點。Python腳本如下:
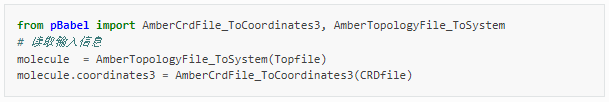
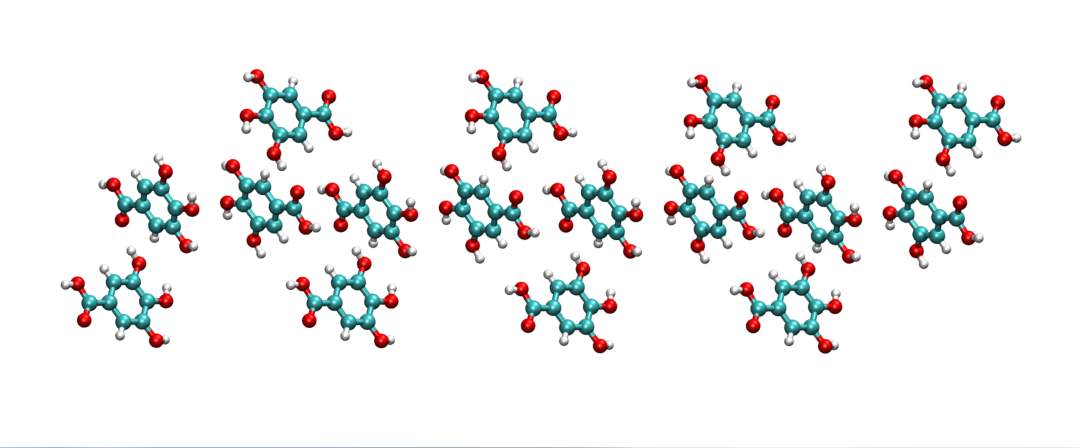
其中,需要提前安裝好AmberTools,python2.0版本,并正確設置好AMBERHOME和PDYNAMO環境變量,關于如何將GallicAcid.pdb初始結構文件(圖1,晶胞為2*1*1)生成使用AmberTools21程序相對應的坐標文件GallicAcid.crd和參數/拓撲文件GallicAcid.prmop的方法如下:
運行antechamber程序將Pdb文件轉化為mol2文件:antechamber -i GallicAcid.pdb -fi pdb -o GallicAcid.mol2 -fo mol2 -j 5 -at amber -dr no
-i 指定輸入文件
-fi 指定輸入文件類型
-o 指定輸出文件
-fo 指定輸出文件類型
-j匹配原子類型和鍵類型
-at定義原子類型
運行parmchk2程序生成對應體系的力場參數文件:parmchk2 -i GallicAcid.mol2 -f mol2 -o GallicAcid.frcmod
運行tleap程序構建系統拓撲并為分子定義力場參數步驟如下:
1.使用tleap命令啟動tleap程序

2. 確定并加載體系力場:source leaprc.gaff(此為GAFF力場)
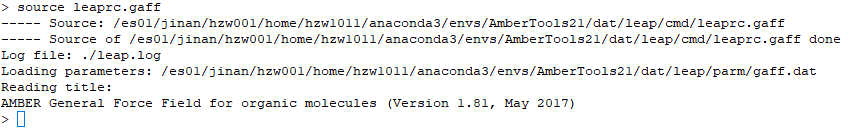
3. 調入配體mol2文件:GA = loadmol2 GallicAcid.mol2

4. 檢查導入的結構是否準確或缺失參數:check GA
5. 調入體系分子的模板,并補全庫文件中缺失的參數:loadamberparams GallicAcid.frcmod
6. 準備生成的Sustiva庫文件:saveoff GA GallicAcid.lib
7. 修改生成的Sustiva庫文件并調入該文件:loadoff GallicAcid.lib

8. 保存.crd和.prmop文件:saveamberparm GA GallicAcid.prmtop GallicAcid.crd

9. 退出tleap程序:quit
分子動力學模擬
1.此處采用amber軟件進行分子動力學模擬,首先對體系進行能量最小化模擬,輸入文件min.in如下:
Initial minimisation of GallicAcid complex
&cntrl
imin=1, maxcyc=200, ncyc=50,
cut=16, ntb=0, igb=1,
&end
imin=1:運行能量最小化
maxcyc=200:能量最小化的最大循環數
ncyc=50:最初的0到ncyc循環使用最速下降算法, 此后的ncyc到maxcyc循環切換到共軛梯度算法
cut=16:以埃為單位的非鍵截斷距離
ntb=0:關閉周期性邊界條件
igb=1:Born模型
使用如下命令運行能量最小化:
sander -O -i min.in -o GallicAcid_min.out -p GallicAcid.prmtop -c GallicAcid.crd -r GallicAcid_min.rst &
其中GallicAcid_min.rst為輸出包含坐標和速度的重啟文件
2.接下來利用最小化模擬得到的重啟文件升溫系統,從而完成分子動力學模擬,輸入文件md.in如下:
Initial MD equilibration
&cntrl
imin=0, irest=0,
nstlim=1000,dt=0.001, ntc=1,
ntpr=20, ntwx=20,
cut=16, ntb=0, igb=1,
ntt=3, gamma_ln=1.0,
tempi=0.0, temp0=300.0,
&end
imin=0:進行分子動力學(MD)
irest=0:讀取先前保存的重新啟動文件讀取坐標和速度
nstlim=1000:運行的MD步數
dt=0.001:時間步長(單位:ps)
ntc=1:不啟用SHAKE約束
ntpr=20:每ntpr步輸出能量信息mdout一次
ntwx=20:每ntwx步輸出Amber軌跡文件mdcrd一次
ntt=3:Langevin恒溫器控制溫度
gamma_ln=1.0:Langevin恒溫器的碰撞頻率
tempi=0.0:模擬的初始溫度
temp0=300.0:模擬的最終溫度
使用如下命令運行分子動力學模擬:
sander -O -i md.in -o md.out -p GallicAcid.prmtop -c GallicAcid_min.rst -r GallicAcid_md.rst -x GallicAcid_md.mdcrd -inf GallicAcid_md.mdinfo
其中GallicAcid_md.mdcrd文件即為MD模擬的軌跡文件,可借助VMD軟件進行可視化顯示分子結構,并從動力學模擬軌跡提取感興趣的結構存儲于.crd文件中。
QM/MM總能量計算
分子動力學模擬后提取文件為GallicAcid.prmtop, GallicAcid.crd,可對體系進行全量子化學總能量計算,python代碼如下:
import glob, math, os
from pBabel import AmberCrdFile_ToCoordinates3, AmberTopologyFile_ToSystem
from pCore import logFile
from pMolecule import QCModelBDF, System
# 讀取水盒子坐標和拓撲信息
molecule = AmberTopologyFile_ToSystem ("GallicAcid.prmtop")
molecule.coordinates3 = AmberCrdFile_ToCoordinates3("GallicAcid.crd")
# 定義能量計算模式,此處為全體系密度泛函計算,可以定義方法和基組,分別為GB3LYP和6-31g,
model = QCModelBDF("GB3LYP:6-31g")
molecule.DefineQCModel(model)
molecule.Summary() #輸出體系計算設置信息
# 計算總能量
energy = molecule.Energy()
除了可以用全量子化學QM計算體系總能量,也可對感興趣的分子進行QM/MM計算(本例為指定第五個分子用QM方法計算),QM/MM組合能量計算python腳本如下:
import glob, math, os
from pBabel import AmberCrdFile_ToCoordinates3, AmberTopologyFile_ToSystem
from pCore import logFile, Selection
from pMolecule import NBModelORCA, QCModelBDF, System
# 定義能量計算模式
nbModel = NBModelORCA() #處理QM和MM區相互作用
qcModel = QCModelBDF("GB3LYP:6-31g")
# 讀取體系坐標和拓撲信息
molecule = AmberTopologyFile_ToSystem("GallicAcid.prmtop")
molecule.coordinates3 = AmberCrdFile_ToCoordinates3("GallicAcid.crd")
# 關閉體系對稱性
molecule.DefineSymmetry(crystalClass = None) #QM/MM方法不支持使用周期性邊界條件,故關閉周期性邊界條件
# 指定QM區
qm_area = Selection.FromIterable(range (72, 90)) # 指定第五個分子用QM方法計算,其中(72, 90)指明原子列表索引值為72,73,74…..87,88,89,該值=原子序數-1
# 定義能量計算模式
molecule.DefineQCModel (qcModel, qcSelection = qm_area)
molecule.DefineNBModel (nbModel)
molecule.Summary()
# 計算總能量
energy = molecule.Energy()
QM/MM模擬的輸出總結了MM部分,QM部分,QM區和MM區相互作用部分的計算細節如下:

輸出體系總能量信息以及各部分能量貢獻如下:
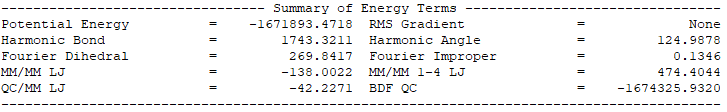
-
軟件
+關注
關注
69文章
4968瀏覽量
87701 -
程序
+關注
關注
117文章
3791瀏覽量
81156 -
動力學
+關注
關注
0文章
105瀏覽量
16984
原文標題:鴻之微BDF軟件計算賞析|采用BDF的QM/MM多尺度計算方法研究晶體的光物理性質
文章出處:【微信號:hzwtech,微信公眾號:鴻之微】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
【一文看懂】什么是量子計算?

一種高靈敏的化學檢測系統
量子通信與量子計算的關系
菱形石墨烯結構及其中的量子反常霍爾效應
華為公開量子計算新專利
“本源悟空”超導量子計算機助力大規模流體動力學量子計算

基于量子干涉技術的單分子晶體管問世
量子計算機應用-量子計算流體力學軟件






 工商網監
工商網監
評論