 有什么方法可以調節界面氫鍵環境提高HOR/HER活性呢
有什么方法可以調節界面氫鍵環境提高HOR/HER活性呢
01 導讀
在全球從化石燃料向可持續經濟轉型的過程中,有效地將可再生能源儲存到氫分子中并將其釋放的重要性,使氫氧化和析出反應(HOR/HER)被賦予了新的意義。開發用于HOR/HER的高效催化系統對加快世界向可再生能源過渡起著至關重要的作用。盡管經過了幾十年的研究,對提供HOR/HER活性的關鍵材料性能的理解仍然不完全。近年來,越來越多的人認識到,HOR/HER活性取決于電化學界面的性質,而不僅僅由催化劑的組成和結構來決定。
02 成果背景
近日Angew. Chem. Int. Ed.期刊上發表了一篇題為“Enhancing Hydrogen Oxidation and Evolution Kinetics by Tuning Interfacial Hydrogen-Bonding Environment on Functionalized Pt Surface”的文章,該工作通過原位表面增強紅外和拉曼光譜證明了特定的吸附有機添加劑(茶堿衍生物)可以通過引入弱氫鍵水將多晶Pt上的本征HOR/HER活性提高3倍。在7-n-丁基茶堿修飾的Pt表面能夠在不消耗界面水分的情況下,充分破壞雙層氫鍵網絡,從而實現最佳的HOR/HER活性。這項工作表明了電化學界面工程作為提高電催化性能策略的前景。
03 關鍵創新
(1)Pt的HOR/HER活性極易受7-取代茶堿的烷基鏈長度的影響,其中弱氫鍵水在活性增強中的關鍵作用,吸附物旁的Pt位點被認為是具有增強的HOR/HER活性的位點。
(2)本文表明界面結構的設計能夠對HOR/HER動力學產生實質性的影響。
04 核心內容解讀
為了了解咖啡因在堿中對HOR/HER動力學起促進作用的源頭,作者采用了與咖啡因結構部分相似的底物,以消除結構基序和/或單個官能團所帶來的影響。咖啡因分子有兩個主要的結構成分(圖1),即1,3-二甲基尿嘧啶和1-甲基咪唑。此外,眾所周知的咖啡因上的甲基會影響其緩蝕劑和藥物配方的性能,因此,咖啡因分子的部分或完全去甲基化的產物(茶堿和黃嘌呤),也會影響HOR/HER活性。
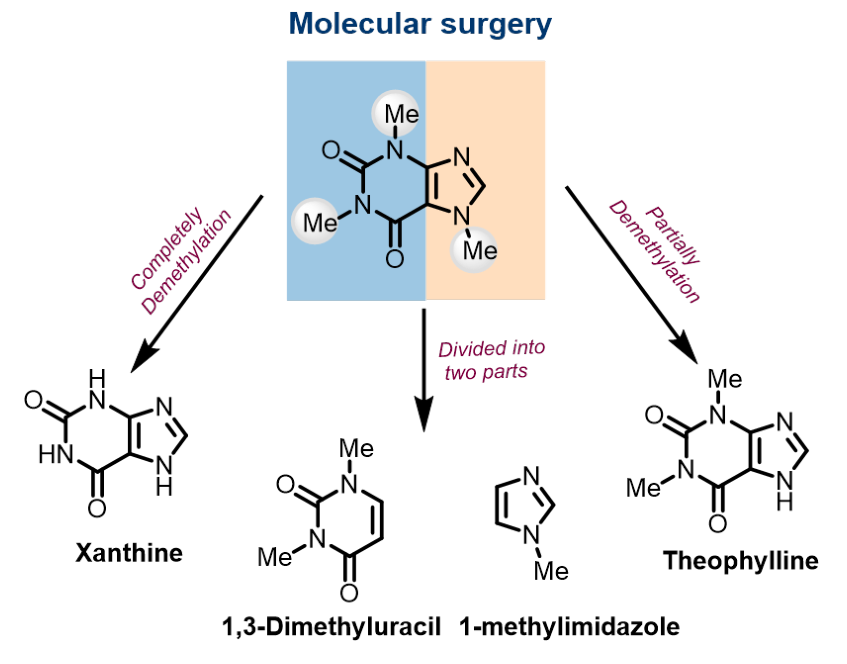
圖1.模擬咖啡因分子片段的分子。@Wiley
本文證明了多晶(pc)Pt上低電位解吸H(Hupd)的循環伏安(CV)特征會被添加的有機物種抑制。在0.1 M的KOH中,0.28和0.38 V處(圖2a)的Pt上存在Hupd解吸的明確CV特征,這分別歸因于pc Pt表面上的110和100面。在電解液中加入0.1 mM的咖啡因(綠色)后,110和100兩個切面上的Hupd特征大多被抑制,這與之前的報道一致。
與此同時,111面上的寬Hupd特征仍占相當大的比例(~65%)。以往的研究表明,芳香族化合物通常在梯田上吸附在Pt上,有機吸附物之間的空間相互作用可以阻止它們形成緊密的填充層,即使在有機吸附物的飽和覆蓋下,也會留下一部分表面位點。110和100兩個面的Hupd特征被有效抑制可能是兩個原因:
1)Pt(110)和Pt(100)上的配位數較低,更有利于咖啡因的吸附,而Pt(111)上的配位數較低,有利于更緊密的吸附構型;
2)pc Pt上Pt(110)和Pt(100)位點的Hupd特征與連接Pt(111)的階梯位點相對應。在這些窄而強的吸附區域中,吸附物有更多的構型自由度來最大化填充,因為在相鄰的階梯位點上,與弱吸附的咖啡因增加的空間相互作用會導致相對較小的能量損失。本文還表明,在0.1 mM的1,3-二甲基尿嘧啶(紅色)和1-甲基咪唑(藍色)存在下,CVs對Hupd特征的抑制分別比咖啡因(圖2a)更少和更多。
與0.1 M KOH相比,電解液中與1,3-二甲基尿嘧啶的Hupd特征更弱、更廣,并轉移到較低的電位,但比咖啡因更明顯(紅色,圖2a)。1,3-二甲基尿嘧啶上的兩個極性羰基可能通過氫鍵增加其對界面水的親和力,這會降低其吸附能和表面覆蓋,而1-甲基咪唑則更疏水,傾向于與Pt表面發生強烈的相互作用。
根據Hupd特征比較,咖啡因和1,3-二甲基尿嘧啶(或1-甲基咪唑)的覆蓋范圍會因為它們不同的表面足跡而變得復雜。同時,從1-甲基咪唑和1,3-二甲基尿嘧啶的相對表面親和力可以推斷,1-甲基咪唑對咖啡因在鉑上的吸附作用更大。
此外,本文還用Butler-Volmer方程擬合極化曲線(圖2b),測定有無有機添加劑的Pt電極固有的HOR/HER活性,來證明有機添加劑對Pt的Hupd特性影響與HOR/HER本征活性無關。
本研究報告的交換電流密度是根據各自有機添加劑在0.1 M的KOH中Hupd峰的面積,用幾何表面積和測量的電化學表面積(ECSA)對Pt進行歸一化得到的(圖2c),它們通常表現出類似的趨勢。
咖啡因和1-甲基咪唑的交換電流密度都比沒有任何有機添加劑時更高,而表面上的1,3-二甲基尿嘧啶的存在略微降低了活性(圖2c)。1-甲基咪唑(無論是單獨使用還是作為咖啡因中的一個片段)增強HOR/HER活性的能力似乎與其與表面的強結合有關(圖2a)。此外,這些結果表明,Hupd特征的強度與HOR/HER活性沒有太強的相關性。在不同的面上,出現Hupd特征的電位與氫結合能(HBE)有關。
由于Volmer步驟通常被認為是HOR和HER中的速率控制步驟(RDS),因此HBE被廣泛用于描述貴金屬催化劑上的HOR/HER活性,即低HBE提供高活性。這種相關性在不同的有機添加劑中不成立,因為與1,3-二甲基尿嘧啶的Hupd特征向較低的電位移動,意味著較低的HBE,而交換電流密度比沒有任何有機添加劑的情況下稍低。
同樣,與未添加任何添加劑的電解質相比,添加了咖啡因和1,3-二甲基尿嘧啶的電解質的OH吸附峰發生了正位移,表明吸附的有機物質削弱了OH在Pt上的結合能(OHBE)。在CV掃描的電位范圍內,1-甲基咪唑的OH吸附/解吸特征基本不存在,表明OHBE進一步減弱。與文獻報道相反,OHBE的趨勢似乎與當前系統中測量的活動沒有任何明確的相關性。
由此可以推斷,HBE和OHBE都可能是影響HOR/HER中催化劑性能的因素,而不是主導因素。
本文證實去除咖啡因中的甲基降低了HOR/HER動力學,表明甲基在反應中不是旁觀者。咖啡因逐漸去甲基化為茶堿和黃嘌呤,導致Hupd特征部分恢復(圖2d)。
DFT計算顯示,茶堿和黃嘌呤中所有N-H鍵的pKa值顯著小于13,表明所有N-H鍵在電解質中都被去質子化。這并不奇怪,因為強的吸電子基團,如羰基,傾向于使相鄰的N-H鍵更酸性,而負電荷被大的共軛結構穩定下來。用N-H鍵取代咖啡因中的N-CH3,使分子更具極性,增強了其與界面水的親和力,從而減弱了吸附能。重要的是,HOR/HER動力學與分子所具有的N-H鍵數量呈負相關(圖2c),表明甲基有助于反應的進行。
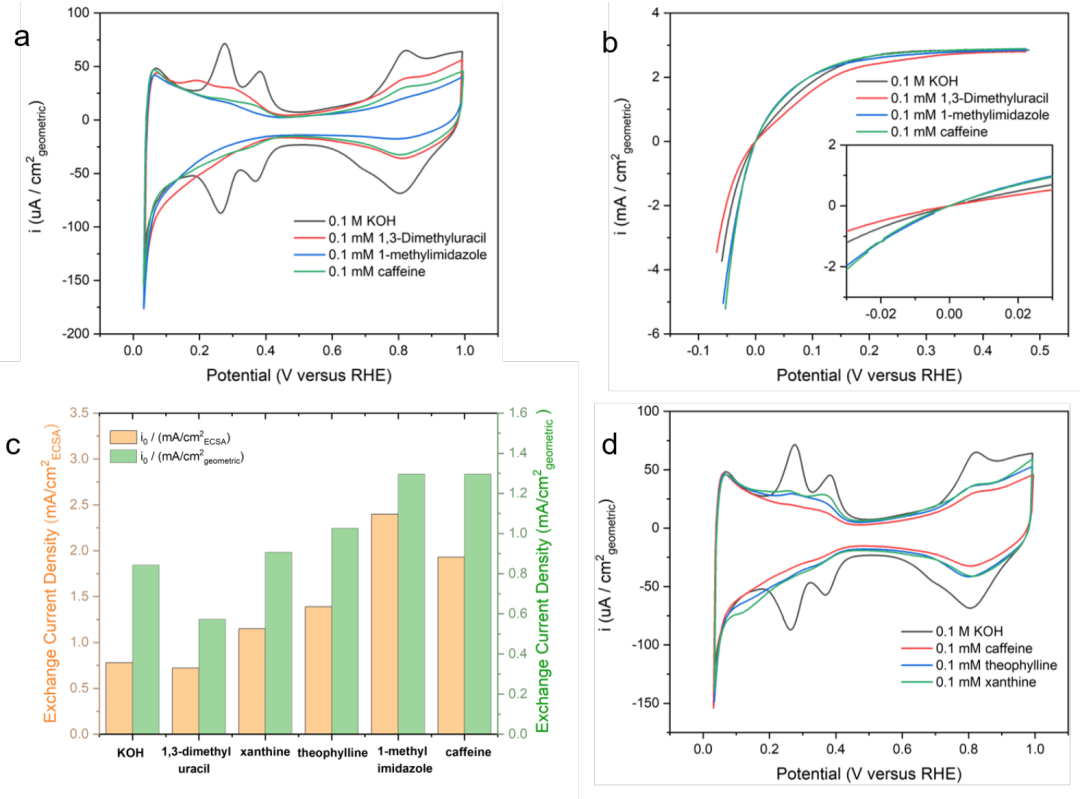
圖2.(a)在Ar飽和的0.1 M的KOH和0.1 M的KOH中加入0.1 mM咖啡因、1,3-二甲基尿嘧啶或1-甲基咪唑的pc Pt上的CV。(b) 0.1 mM咖啡因、1,3-二甲基尿嘧啶或1-甲基咪唑溶液中飽和氫的0.1 M的KOH和純0.1 M的KOH溶液中pc Pt的HOR/HER極化曲線。(c)在氫飽和0.1 M的KOH和0.1 M KOH中使用0.1 mM有機添加劑測量的pc Pt上的HOR/HER交換電流密度。(d)在Ar飽和的0.1 M的KOH和0.1 M KOH中加入0.1 mM咖啡因、茶堿和黃嘌呤對pc Pt的CV。@Wiley
實驗結果表明,Pt的HOR/HER活性極易受7-取代茶堿的烷基鏈長度的影響。由于咖啡因中的甲基對HOR/HER動力學有積極的影響,作者假設吸附質的疏水性可能是優化活性的一個關鍵參數,進而合成了一系列含0~6個碳原子烷基鏈的7-取代茶堿。所有7-取代茶堿衍生物的CV在電流密度和形狀上相似,表明覆蓋范圍相似。
重要的是,HOR/HER的交換電流密度隨著烷基鏈長度增加到4個碳原子而增加,然后趨于穩定,并隨著進一步增加而下降(圖3a)。在7-正丁基茶堿修飾的Pt表面上,HOR/HER的交換電流密度比未修飾的pc Pt表面高約3倍。7-異丙基茶堿(圖3a中藍色菱形)在0.1 mM時的HOR/HER活性比7-正丙基茶堿低約30%,表明烷基的結構以及碳原子數量影響活性。
經過表面修飾后的7-(4,4,4-三氟)丙基茶堿(圖3a中綠色的圓圈)活性也比7-正丁基茶堿差。
7-取代茶堿衍生物的電化學阻抗譜(EIS)進一步支持了它們對HOR/HER活性的影響。200 mV時的EIS光譜中出現了兩個半圓,低頻時的左半圓與氫吸附過程的電荷轉移電阻(Rct)有關,右半圓與雙層電容有關。在EIS中測定的Rct與非均相法拉第反應的動力學成反比。
在本研究中,反映了氫吸附和解吸反應的動力學,即H2O + e-Had+ OH-。隨著7-取代茶堿衍生物上烷基鏈的碳數從1增加到4,200 mV時的Rct逐漸減小,反應壁壘降低(圖3c)。在7-取代茶堿衍生物系列中,測量的隨機對照實驗值和交換電流密度有一個關鍵的差異。7-n-己基茶堿的Rct與7-n-正丁基茶堿相當,而前者在HOR/HER交換電流密度上顯著較低(圖3a)。
這種差異可以歸因于活動和隨機對照試驗測量中采用的不同條件。前者與電流密度在1 mA/cm-2量級的連續電化學反應,而后者則在電流密度低得多的情況下(峰對峰電壓變化為5 mV)對Hupd進行高頻吸附和解吸。因此,活度測量需要通過水和H2的質量輸送來持續提供反應物,而EIS測量是在Hupd區域(200 mV vs. RHE)內進行的,在該區域中,Rct的測定不太可能受到反應物質量輸送限制的影響。
實驗結果表明,7-正丁基茶堿和7-正己基茶堿修飾Pt上的氫吸附/解吸動力學是相似的,但7-正己基茶堿中較長的烷基鏈在HOR/HER活性測試中阻礙了H2和/或水的界面傳質。
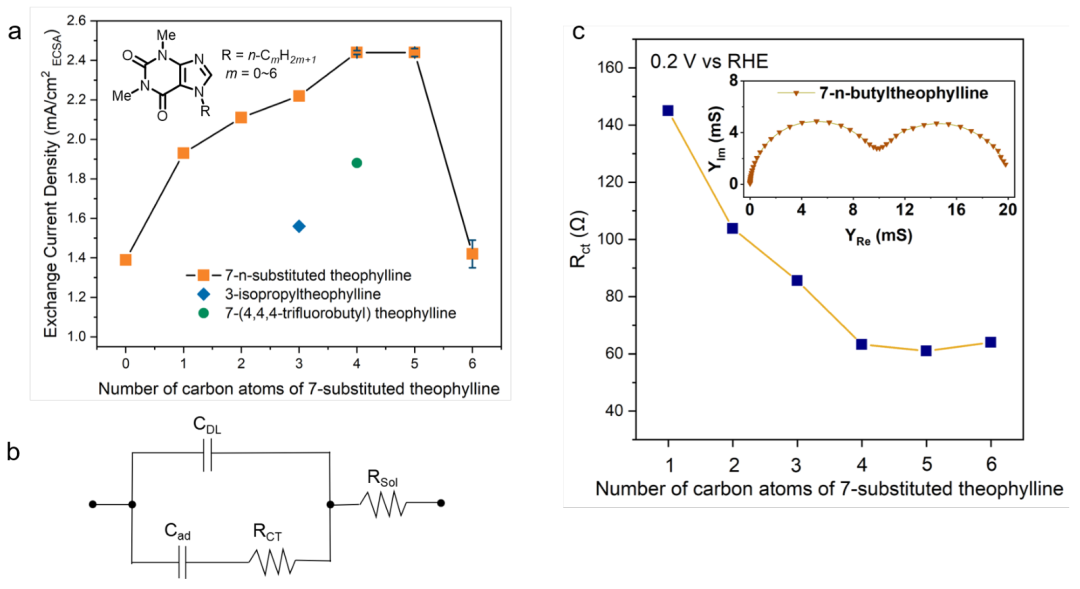
圖3.(a) 0.1 mM的茶堿衍生物在0.1 M的KOH飽和H2下測定pc Pt上的HOR/HER活性。(b) Hupd區等效電路。(c) 電荷轉移電阻與7-取代茶堿碳原子數的函數關系。@Wiley
為了了解這些有機添加劑在鉑表面的吸附性質,對7-正丁基茶堿修飾的鉑表面進行了原位表面增強紅外吸收光譜(SEIRAS)研究。其中重要的是,當電勢階躍降到0 V時,羰基帶紅移到1678 cm-1(圖4a)。振動斯塔克效應是特異性吸附的標志,表明7-正丁基茶堿與金屬表面直接接觸。
本文使用CO作為探針分子來進一步證實7-正丁基茶堿的特定吸附這一機理。CO在鉑表面的吸附力很強,可以取代其他弱結合的吸附物,如水。CO引入到7-正丁基茶堿覆蓋的表面后,在2099、2031和1826 cm-1處出現了峰,其中前兩峰可以被不同類型位點線性連接的CO匹配,后一條可以被橋接連接的CO匹配。
CO共吸附后,羰基峰的強度明顯下降,但仍保留了其大部分強度,表明CO不能完全取代被吸附的7-正丁基茶堿。
在CO共吸附和不共吸附的情況下,羰基模式也觀察到了類似的斯塔克振動效應(圖4b),進一步證實了7-正丁基茶堿在Pt表面的特異性吸附。結果表明,7-正丁基茶堿在Pt上的吸附能與CO的吸附能相當,這與CV中Hupd和OH吸附/解吸區特征的抑制相吻合。
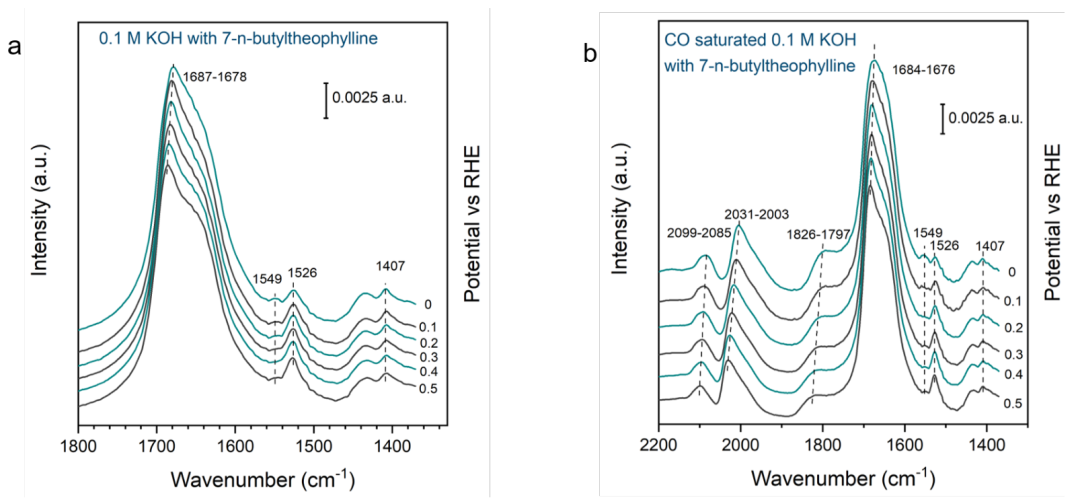
圖4.(a) Ar氣氛下,0.1 mM的7-正丁基茶堿在Pt膜電極上0.1 M的KOH中的電位相關原位SEIRA光譜。(b) CO氣氛中,0.1 mM的7-正丁基茶堿在Pt上0.1 M的KOH中的電位相關原位SEIRA光譜。@Wiley
電化學界面上弱氫鍵水的比例與HOR/HER反應動力學密切相關。在0.2到-0.1 V的電位范圍內,0.1 M的KOH范圍內可以看到從3000 cm-1到3800 cm-1的寬頻峰,這通常歸因于水的各種O-H拉伸模式。
根據廣泛的O-H伸縮帶的反褶積,文獻提出了三種不同的水的配位環境:~3600 cm-1弱H鍵水分子、~3400 cm-1不對稱H鍵水分子(陽離子水合層中的水)和3200 cm-1對稱H鍵水分子(冰狀水或體積水)。
7-正丁基茶堿在Pt電極上吸附后,在0.2 V時在3587 cm-1處出現一個尖銳的峰,該峰位于弱H鍵水的區域(圖5a)。這個峰隨著電位的降低而紅移,觀察到的振動斯塔克效應表明,產生這個峰的水分子位于電極表面附近。這個銳利的峰出現在所有吸附了7-取代茶堿衍生物的Pt表面(圖5b)。
重要的是,相對于主要的寬O-H拉伸帶,這一明顯特征的強度隨著這些有機添加劑的7位烷基鏈的碳數增加而增加,直到7-正丁基茶堿變緩,后開始下降直到7-正己基茶堿,這與HOR/HER交換電流密度的趨勢一致。
結果表明,吸附在7-取代茶堿上的烷基可以破壞界面水的氫鍵網絡,從而降低水的活化能。界面水分子H鍵的弱化使得改變其構型以穩定HOR/HER激活的配合物的能量成本更低,即破壞現有配位環境的能量損失更低。
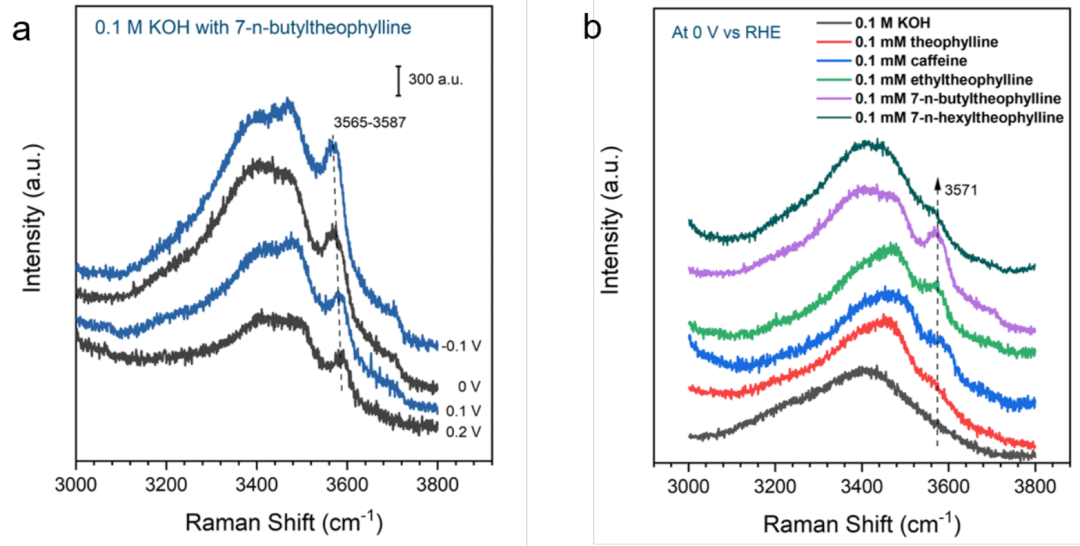
圖5.在0.1 M的KOH溶液中,(a)加入0.1 mM 7-正丁基茶堿在-0.1~0.2 V的界面水的pc Pt電極上的拉曼光譜,(b)加入0.1 mM不同茶堿衍生物在0 V的界面水的pc Pt電極上拉曼光譜。@Wiley
05 成果啟示
綜上所述,7-n-烷基取代茶堿衍生物的HOR/HER活性與烷基鏈長度呈火山形狀關系,最佳烷基鏈碳數n為4。原位SEIRAS實驗表明,7-正丁基茶堿在Pt表面特異性吸附,而原位拉曼光譜顯示出明顯的特征,對應于茶堿衍生物修飾Pt上弱氫鍵水。
隨著烷基鏈碳數從1增加到4,拉曼特征強度增加,然后進一步增加到6,拉曼特征強度下降。這一趨勢與HOR/HER交換電流密度的趨勢相一致,表明弱氫鍵水在活性增強中的關鍵作用。
吸附物旁的Pt位點被認為是具有增強的HOR/HER活性的位點。該工作表明界面結構對HOR/HER動力學有實質性的影響,并強調了工程界面微環境作為增強電催化反應策略的前景。
審核編輯:劉清
-
電阻
+關注
關注
86文章
5507瀏覽量
171931 -
電解質
+關注
關注
6文章
810瀏覽量
20049 -
電荷
+關注
關注
1文章
628瀏覽量
36134
原文標題:Angew: 調節界面氫鍵環境提高HOR/HER活性
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
如何提高信噪比的有效方法

放大器增益的調節方法
THS4524 CMRR想提高到140dB及以上,請問有好的方法嗎?
人機界面應具備的特性是什么
溫度繼電器的調節方法有哪些
變頻器壓力調節最簡單方法是什么
AWTK 開源串口屏開發(14) - 界面重用






 工商網監
工商網監
評論