

【研究背景】
極限快速充電(XFC),即在10~15分鐘內獲得80%電池容量的快速充電標準,是當今時代對于電池快充提出的新要求。然而,受限于電池本征的極化現象,大多數電池難以達到此標準。對此,人們曾提出自加熱結構設計提升電池內部的反應動力學,但是高溫不可避免會加劇體系副反應,電解液分解和電極/電解質界面穩定性往往是相互關聯的,這很大程度上進一步阻礙了快充的實現。
合理的溶劑化結構重組有助于降低金屬離子的去溶劑化能壘,從而使電極/電解質界面上的電荷轉移動力學更快。經過這樣的電解質溶劑化設計和SEI調整,電池的循環穩定性得到了改善。然而,這些改進是否可以在熱調制下保持尚不清楚。在快速充電電池中,利用定制溶劑調節界面化學往往有著的奇妙的作用。
【成果簡介】
近日,哈工大王振波教授、趙磊課題組與華僑大學闕蘭芳團隊在Angew. Chem. Int. Ed.上發表題為“Constructing Stable Anion-Tuned Electrode/Electrolyte Interphase on High-Voltage Na3V2(PO4)2F3 Cathode for Thermally-Modulated Fast Charging Batteries”的研究論文。本文研究發現鈉電體系中,電解質溶劑化結構中的陰離子/溶劑比越高,形成的界面相中陰離子占主導,包含更多的無機離子和更多的陰離子衍生物(CxClOy),這會阻礙Na+快速傳輸。相比之下,較低的陰離子/溶劑比可獲得穩定的陰離子調節界面相,從而獲得更好的界面動力學和循環能力。該工作對于快充高壓鈉電體系的設計具有重要重要意義。
【研究亮點】
(1)研究了高壓Na3V2(PO4)2F3正極在有機碳酸鹽溶劑中充電至4.5 V、55℃時,不同濃度NaClO4組成的電解質之間形成的界面相,發現電解質中陰離子/溶劑比越高,形成的界面相中陰離子占主導,愈發不利于Na+的快速傳導。
(2)研究發現,較低的陰離子/溶劑比例設置可獲得穩定的陰離子調節界面相,從而獲得更好的界面動力學和循環能力。同時,通過觸發CxClOy的分解,可以補償正極材料的性能衰退,恢復電池性能。
【圖文導讀】
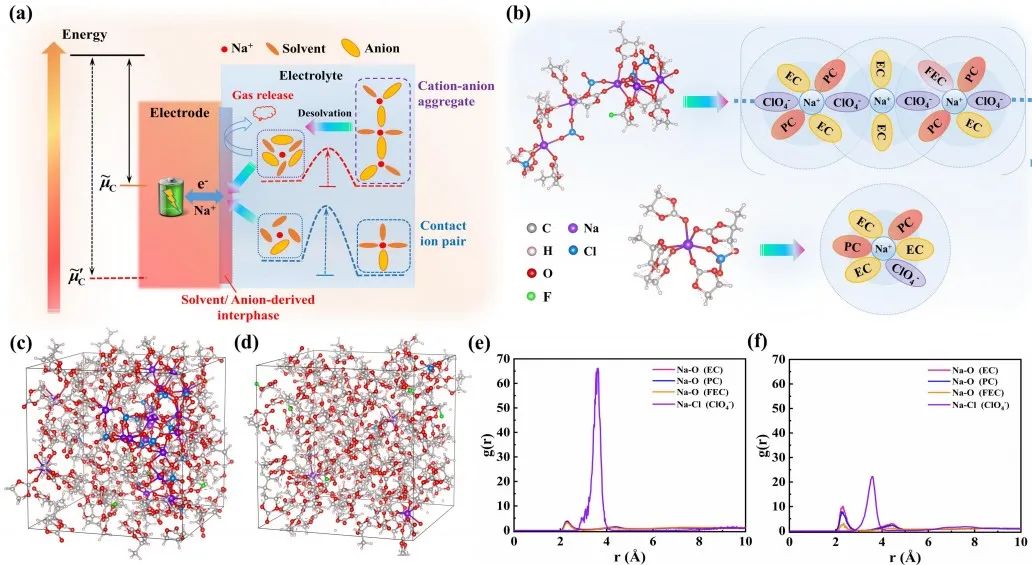
圖1 電解質的溶劑化結構。(a) 電極/電解質界面示意圖;(b)長鏈AGGs在HCE、LCE和ULCE中可能的溶劑化構型;(c) HCE的MD模擬結果;(d) ULCE的MD模擬結果;(e) HCE的Na+徑向分布函數MD模擬結果; (f) ULCE的Na+徑向分布函數MD模擬結果。
如圖1a所示,當電極與電解質接觸時,受到彼此吉布斯自由能的熱力學差異作用,反應產物則成為了電極/電解質界面相的組成部分。兩種電解質的典型溶劑化結構如圖1b所示,MD模擬結果顯示(圖1c和d)顯示,隨著濃度的增加,溶劑化絡合物在HCE和LCE中傾向于形成長鏈AGGs,而在ULCE中分離的CIPs則呈現自定型結構。結合徑向分布函數(圖1e和f),這些結果表明,ClO4-陰離子通過與更多的Na+絡合形成重復的AGG單元而主導溶劑化鞘組成結構。
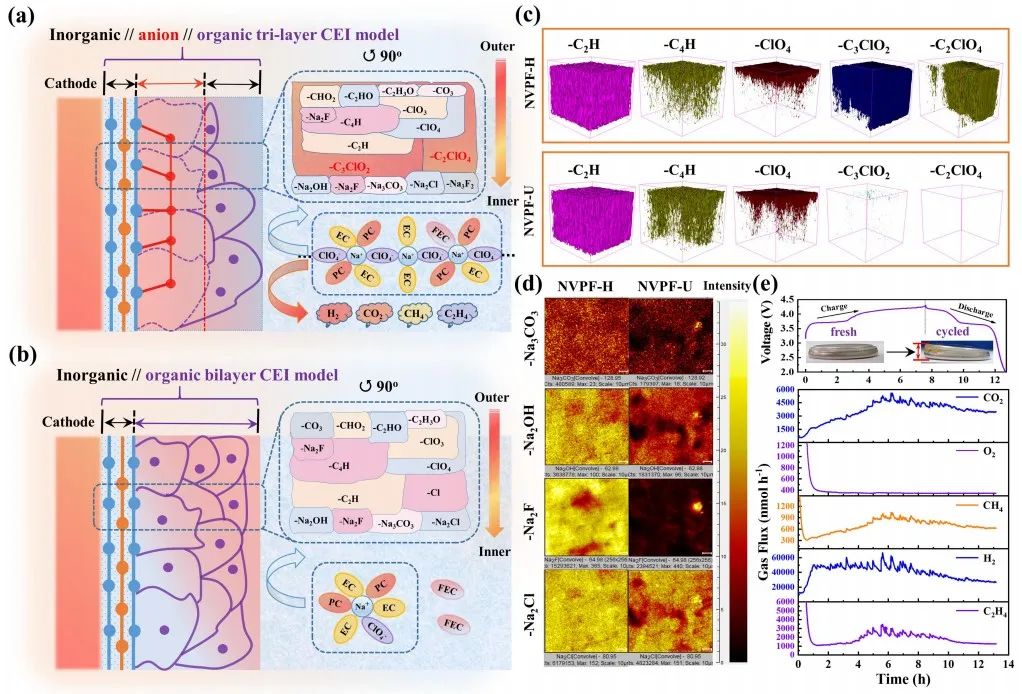
圖2 相間組分及分布。(a) NVPF-H上衍生界面相的結構示意圖;(b) NVPF-U上衍生界面相的結構示意圖;(c) NVPF正極上衍生界面相中一些代表性離子的空間分布;(d) NVPF正極上衍生界面相中代表性無機物質的TOF-SIMS二維映射圖像;(e) 原位DEMS揭示了NVPF-H體系在55℃第一個循環期間的氣體產生過程。
為了更好地說明兩者之間的區別,根據建立好的體系結構和分布,對溶劑-陰離子衍生界面相的結構組分和分布信息進行了有機//無機多層界面相建模,如圖2a-2b所示。其理論結構示意圖只代表了界面相在某一時刻的狀態,而不是整個循環過程。
如圖2c和2d所示,與HCE (NVPF-H)相比,ULCE (NVPF-U)衍生的界面相中有機組分(RONa, RCOONa, ROCO2Na, R代表烷基)占比較大,無機組分(Na2CO3, NaF, NaCl, NaOH)占比較小。結合上述對溶劑化鞘層的分析,這種差異可以解釋為溶劑化鞘層中溶劑比例較高,有助于溶劑主導的界面相的形成。
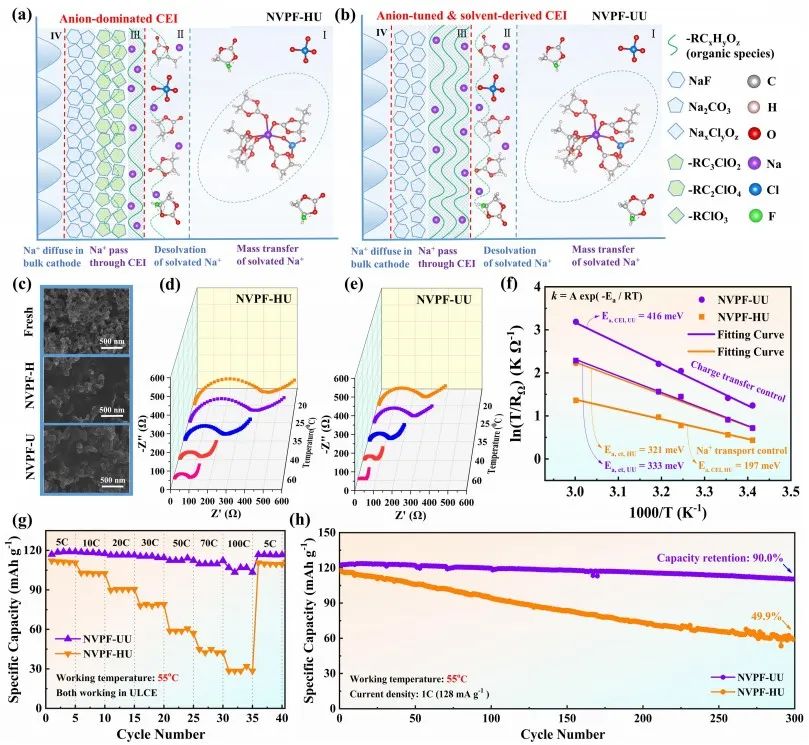
圖3 界面過程和動力學。(a-b) NVPF-HU和NVPF-UU系統中電極/電解質界面示意圖;(c) 經過10次活化循環的NVPF-H和NVPF-U正極的SEM 圖;(d-e) NVPF-HU和NVPF-UU的Nyquist圖;(f) NVPF-HU和NVPF-UU體系中Na+通過CEI和去溶劑化過程的Arrhenius圖和計算活化能;(g) NVPF-HU和NVPF-UU的倍率性能;(h) 在55℃下,NVPF-HU和NVPF-UU的循環穩定性測試。
為了評價陰離子調節對界面反應和動力學的影響,作者進行了控制電極和電解質的單變量對比實驗。如圖3a-3b所示,將新鮮的NVPF電極組裝到電池中,首先分別在HCE、LCE和ULCE中形成界面相。然后,激活的電極被重新組裝成新的電池,并充滿ULCE。
SEM圖像和XPS刻蝕結果表明,該操作后,如圖3c所示,NVPF正極表面形成了界面產物(包括-CxClOy, RCO2Na等)。為了說明這一點,作者使用配備了三電極單元的溫度控制EIS來研究電極/電解質界面的界面過程,如圖3d-e。由于考慮到Na+去溶劑化過程幾乎相同,因此兩個電極的電荷轉移阻抗(Rct)差異很小。而NVPF-HU的Na+界面膜阻抗(RCEI)阻抗幾乎是NVPF-UU的兩倍,表明其Na+輸運動力學受到阻礙。
但是,結合Arrhenius定律計算活化能的結果表明,NVPF-HU的界面離子轉移能壘低于NVPF-UU,這意味著其離子輸運過程在能量上是有利的(圖3f)。這兩點看似相互矛盾,但倍率和循環穩定性測試結果證實,NVPF-HU、NVPF-HL和NVPF-HH的界面Na+輸運動力學確實較差(圖3g-h)。
由于構建了穩定的陰離子調節的CEI, NVPF-UU具有更好的界面相容性和界面Na+輸運動力學。因此通過調節功能間相組分,構建穩定的電極/電解質間相,建立快速的界面離子輸運動力學是激活更好的快充電的關鍵。這里,在NVPF-U上形成的陰離子調優和溶劑衍生的界面相就是一個例子。
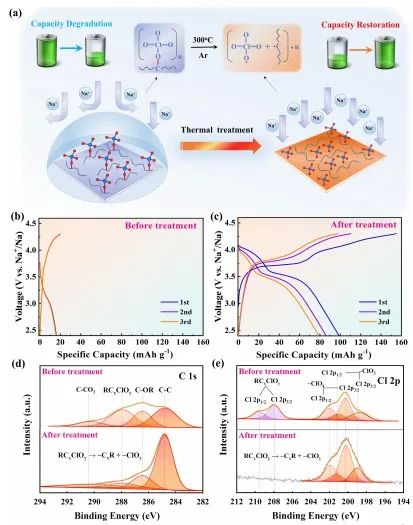
圖4 界面衰退和二次活化。(a)熱處理過程及其對失效NVPF-H正極的影響示意圖;(b)在55℃、1C下,失效NVPF-H正極經過500次循環后的GCD曲線;(c)在55℃、1 C時,經過熱處理的NVPF-H正極的GCD曲線;(d-e)熱處理前后NVPF-H正極的XPS光譜。
為了進一步闡明陰離子調節在界面穩定性和循環耐久性中的作用,作者對失效的NVPF-H電極進行熱重構實驗,如圖4a所示。由于電極/電解質界面不穩定和庫侖效率低,NVPF-H的容量在55 ℃的高溫調制下迅速衰減。在1C下循環500次后,其剩余容量下降到僅約18mAh/g (圖4b)。
有趣的是,失效的NVPF-H在Ar氣氛的保護下經過熱處理后,容量恢復到約100 mAh/g (圖4c)。然而XRD結果顯示,NVPF的晶格結構無明顯變化。
因此,性能的衰減和恢復可能和界面的衰減與重構有關。 XPS結果顯示(圖4d-4e),由于熱處理引發了陰離子衍生物的化學鍵重排(RCxClOy, R代表烷基),NVPF-H界面上C和Cl的化學狀態發生了顯著變化。
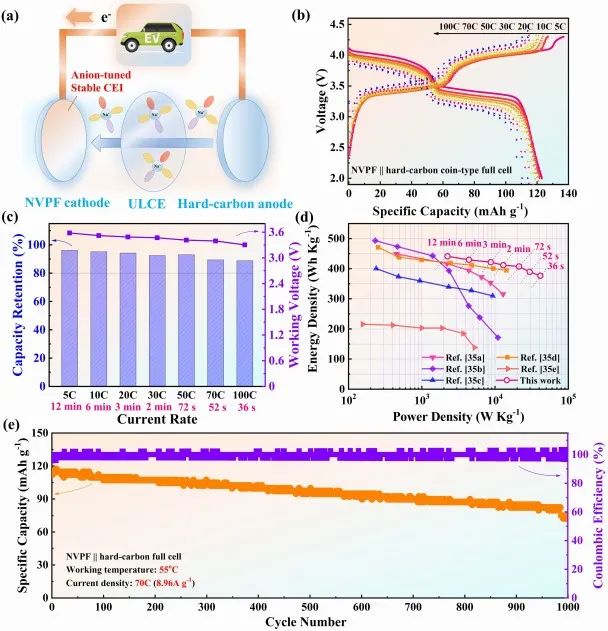
圖5 全電池性能。(a) 電池結構示意圖;(b) 不同倍率下的GCD曲線;(c)不同倍率下全電池的容量保留率(與理論值比較)和平均工作電壓;(d) 性能對比的Ragone圖;(e) 全電池循環穩定性。
為了進一步驗證陰離子調節界面相的效果,組裝Na3V2(PO4)2F3//硬碳全電池,如圖5a所示。其GCD曲線顯示,由于各種極化,當倍率從5 C增加到100 C時,其工作電壓和容量都有較小的下降(圖5b)。但由于在55℃熱調制溫度下電極動力學加速,全電池容量保持在其理論容量的80%以上,工作電壓高于3.3V (圖5c)。
此外,組裝全電池在100 C(36秒)下獲得了其理論容量的88.8%,并提供了376 Wh/Kg的能量密度(基于正活性物質)(圖5d)。此外,整個電池在55℃、70℃下運行1000個循環(900次循環后容量保持率為75.5%)(圖5e)。
【總結和展望】
本工作研究了鈉離子電池中高壓Na3V2(PO4)2F3正極電極/電解質界面相的性質。高濃度電解液中陰離子/溶劑比越高,形成的界面相以陰離子為主,包含更多陰離子衍生物(-CxClOy)和無機物質(Na2CO3、NaF等),導致界面Na+輸運受阻,產氣也越嚴重。
相比之下,在陰離子/溶劑比較低的稀電解質中,接觸離子對的溶劑化構型有助于構建穩定的陰離子調優和溶劑衍生的復合界面相,從而提高在高溫下的性能。
特別地,本工作證明了失效的Na3V2(PO4)2F3正極通過退火可以恢復電極的性能,在Ar下,在300℃下可以分解CxClOy,重建界面相。
在未來的研究中,可能需要優化電解液(特別是電解液濃度和溶劑化結構),以平衡最少的電解液成本和最優的電池性能。
審核編輯:劉清
-
充電電池
+關注
關注
1文章
170瀏覽量
25470 -
電解質
+關注
關注
6文章
821瀏覽量
20425 -
XPS
+關注
關注
0文章
97瀏覽量
12145
原文標題:哈工大王振波Angew:陰離子/溶劑比例調控CEI助力高壓快充鈉電
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦

離子液體添加劑用于高壓無負極鋰金屬電池

鉀離子輔助的多陰離子材料—鈉離子電池長循環穩定性的新機制

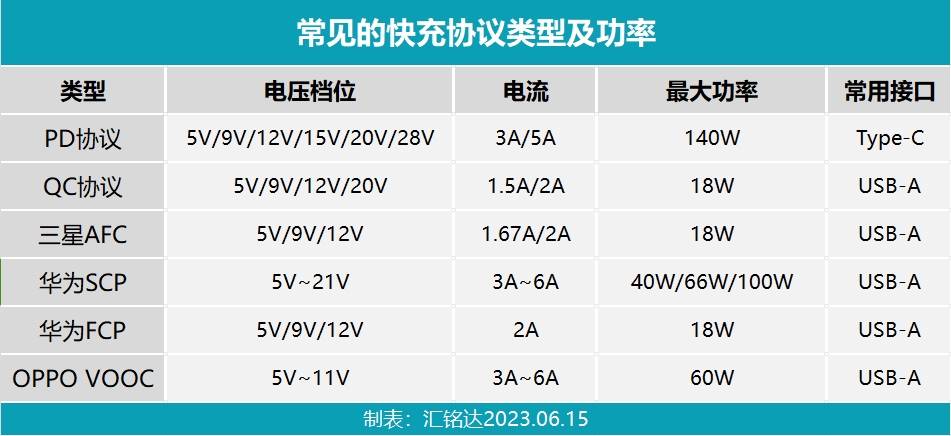
講解什么是取電協議芯片及快充協議的種類
供應智融S621雙向快充移動電源IC
智融SW6201S 支持 PD 的多協議雙向快充移動電源IC
快充工作原理,解讀什么是快充協議及協議芯片的應用



超級快充閃充協議





 工商網監
工商網監

評論