 通過費托合成工藝提高CO加氫對高碳氫化合物的選擇性
通過費托合成工藝提高CO加氫對高碳氫化合物的選擇性
01、研究背景
通過費托合成(FTS)工藝將CO加氫轉變為高附加值化學品已成為學術界和工業界最前沿的研究領域。鎳基催化劑一直被認為是FTS的重要候選材料,與Co或Fe催化的FTS相比,它們可在相對溫和的操作條件下表現出較高的CO加氫活性,但由于羰基鎳的形成,普遍存在的甲烷化和快速失活現象嚴重阻礙了鎳基催化劑的實際應用。因此,開發一種新型的鎳基催化劑,有效地抑制鎳羰基的形成,同時增強C-C偶聯能力,從而提高CO加氫對高碳氫化合物的選擇性,是非常有意義的。
02、成果簡介
鑒于此,北京大學馬丁和王蒙、上海交通大學劉晰、中國科學技術大學劉進勛(共同通訊作者)等人通過對煅燒過的超薄NiTi-LDHs前驅體進行還原,成功合成了具有強烈金屬-載體相互作用(SMSI)型TiO2-x覆層修飾的鎳納米粒子催化劑(TiO2-x/Ni),表現出優異的催化活性。相關成果以“Boosting CO hydrogenation towards C2+hydrocarbons over interfacial TiO2?x/Nicatalysts”為題發表在Nature Communications上。
03、研究亮點
1、以LDHs為前驅體制備了界面TiO2-x/Ni催化劑對C2+烷烴具有~ 19.8 %的高CO轉化率和~64.6 %的C2+烷烴選擇性。
2、TiO2-x/Ni中的Niδ-/TiO2-x界面位點增強了CO的解離并促進了C-C鏈的偶聯。
04、圖文介紹
、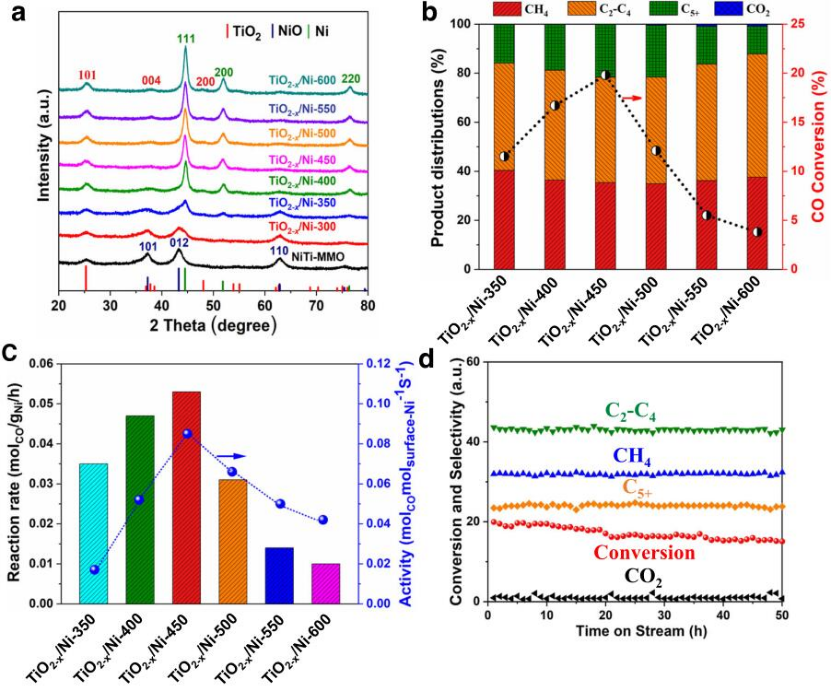
圖1 TiO2-x/Ni催化劑在不同條件下合成氣轉化中的結構性質和催化性能。(a)NiTi-MMO和各種TiO2-x/Ni催化劑在不同溫度下還原的XRD圖譜;(b)各種TiO2-x/Ni催化劑上的轉化率和產物分布以及(c)反應速率和活性;(d)TiO2-x/Ni-450在220℃下的穩定性評價。反應條件:催化劑(120 mg),1 bar,合成氣(CO/H2/Ar=32/64/4;空速: 10000 m L gcat-1h-1)。
采用水熱法合成了NiTi-LDHs納米片。在500℃下煅燒4h后,得到由NiO和TiO2-x組成的混合氧化物(NiTi-MMO)。之后,分別在300℃、350℃、400℃、450℃、500℃、550℃和600℃對NiTi-MMO進行H2還原。還原后的樣品被命名為TiO2-x/Ni-T,其中T表示還原溫度。XRD數據(圖1a)顯示,在350℃還原后觀察到金屬Ni的形成。
在220℃,重時空速(WHSV)為10,000m L gcat-1h-1時,評估了不同溫度下還原的TiO2-x/Ni催化劑的催化性能(圖1b)。產品分布表明,在TiO2-x/Ni催化劑上進行CO加氫時,C2+烷烴是最主要的產品,而不是甲烷(CH4)。六種TiO2-x/NiT催化劑在相同條件下顯示出類似的C2+烷烴選擇性,表明所有的催化劑都含有類似的活性點。各種TiO2-x/Ni催化劑的催化反應速率和活性隨著催化劑還原溫度的增加呈現出類似火山的趨勢(圖1c)。最佳的TiO2-x/Ni-450催化劑在220℃時顯示出最高的反應速率(0.053 molCOgNi-1h-1)和催化活性(0.085 molCOmolsuface-Ni-1s-1)。此外,TiO2-x/Ni-450催化劑在220℃下50h內顯示出良好的穩定性(圖1d)。
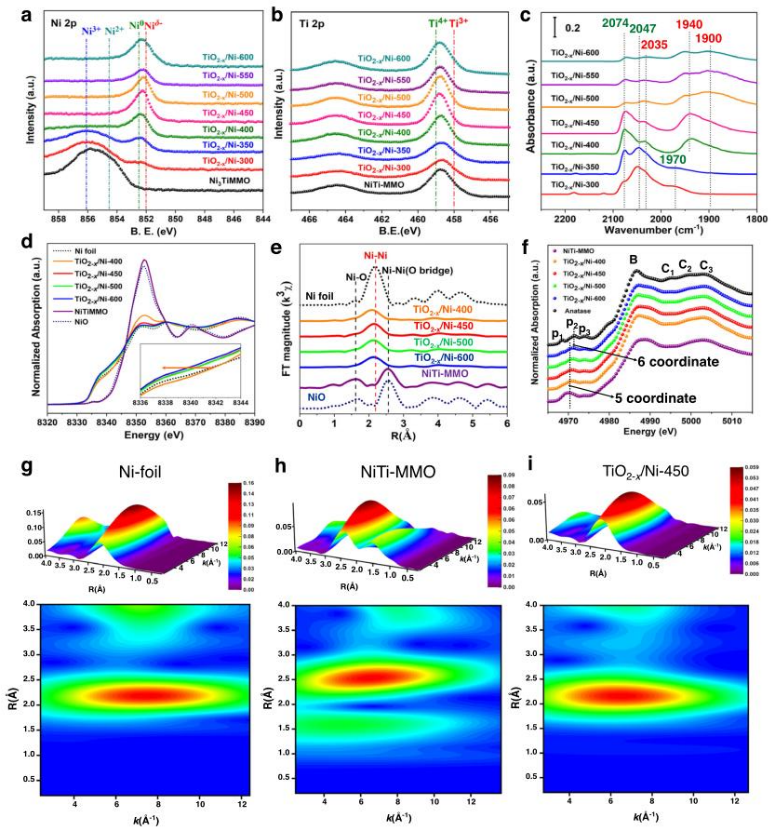
圖2各種TiO2-x/Ni催化劑的幾何結構和電子結構的揭示。各種TiO2-x/Ni催化劑的(a)Ni 2p和(b)Ti 2p的原位XPS譜;(c)TiO2-x/Ni催化劑的室溫原位CO-DRIFTS譜;(d)TiO2-x/Ni催化劑的歸一化Ni K邊XANES譜;(e)相應的歸一化Ni K邊傅里葉變換EXAFS譜;(f)各種TO2-x/Ni催化劑的歸一化Ti K邊XANES譜;(g)Ni箔、(h)NiTi-MMO、(i)TiO2-x/Ni-450催化劑的WT分析。
采用準原位XPS來揭示在不同溫度下激活的各種TiO2-x/Ni樣品的表面電子結構(圖2a,b)。通過煅燒NiTi-LDHs前驅體制備的NiTi-MMO853.4 eV的2p3/2峰為Ni2+物種。由于NiTi-MMO中形成豐富的VNi物種,856.1 eV的峰歸為Ni3+物種,表明Ni3+和Ni2+物種存在于NiTi-MMO中。而在TiO2-x/Ni-300催化劑中,觀察到852.5 eV處有一個微弱的2p3/2峰,為Ni0物種。隨著還原溫度上升到400℃,在852.0 eV處發現一個更強的2p3/2峰,為Niδ-物種。當還原溫度上升到600℃時,位于852.0 eV的2p3/2峰變得更寬更強,表明在進一步還原的催化劑中,Niδ-物種的含量更高。在圖2b中可以觀察到各種TiO2-x/Ni催化劑在458.0 eV處存在Ti3+物種。
為了進一步確認各種TiO2-x/Ni催化劑上存在電子富集的Niδ-物種,進行了原位CO-DRIFTS化學吸附實驗(圖2c)。在25℃時,~2074、~2047和~2035 cm-1的紅外譜帶分別歸屬于鎳缺陷位點、Ni0頂部位點和Niδ-頂部位點上的線性CO分子。當溫度從350℃上升到600℃時,由于TiO2-x亞氧化物的結晶度和覆蓋率提高,這些紅外帶減少,從而抑制羰基鎳的形成。電子富集的Niδ-物種具有更多的d電子,可以加強傳輸電子到2π*反鍵軌道的CO分子,從而加強CO化學吸附。
采用原位XAFS光譜來揭示各種TiO2-x/Ni樣品的電子狀態、幾何結構和配位環境。就NiTi-MMO而言,歸一化的Ni K-邊XANES(圖2d)顯示出較高的白線,并且與NiO參考相比,吸附邊向高光子能量移動,表明NiTi-MMO前體中存在Ni3+物種。前驅體在400℃的10% H2/He氣氛中活化后,主要的相為金屬鎳。結合XPS和CO-DRIFTS表征,表明在TiO2-x/Ni-400、TiO2-x/Ni-450、TiO2-x/Ni-500和TiO2-x/Ni-600催化劑上形成電子富集的Niδ-物種。在圖2e中TiO2-x/Ni在400到600℃的還原溫度范圍內的R空間圖中,四種催化劑的Ni-Ni殼顯示出相似的配位數。Ni箔(圖2g)、NiTi-MMO(圖2h)和TiO2-x/Ni-450(圖2i)的Ni K邊EXAFS振蕩的小波變換(WT)表明NiTi-MMO被還原為金屬顆粒。
為了探索Ti-O的配位環境,進行了原位Ti K-邊XANES。在大于4984 eV處,TiO2-x/Ni-450顯示出比銳鈦礦寬而不明顯的峰,表明Ti-O配位環境嚴重無序的性質(圖2f)。4984eV以下區域,三個峰被標記為P1、P2和P3,是由于核心電子向Ti 3d4p4s軌道的雜化態過渡引起的。在TiO2-x/Ni-450中,與標準銳鈦礦(4972.2 eV)相比,4970.6 eV的P2峰強度明顯增加并轉移到低能量處,此處的峰是由五配位的Ti原子引起的,這與Ov-Ti3+物種的形成密切相關。SMSI可以發生在界面上,伴隨著電子從Ov-Ti3+物種轉移到相鄰的界面Ni原子上,形成Niδ-位點。因此,豐富的Niδ-/TiO2-x界面位點可以在TiO2-x/Ni-450催化劑上有效形成并穩定。
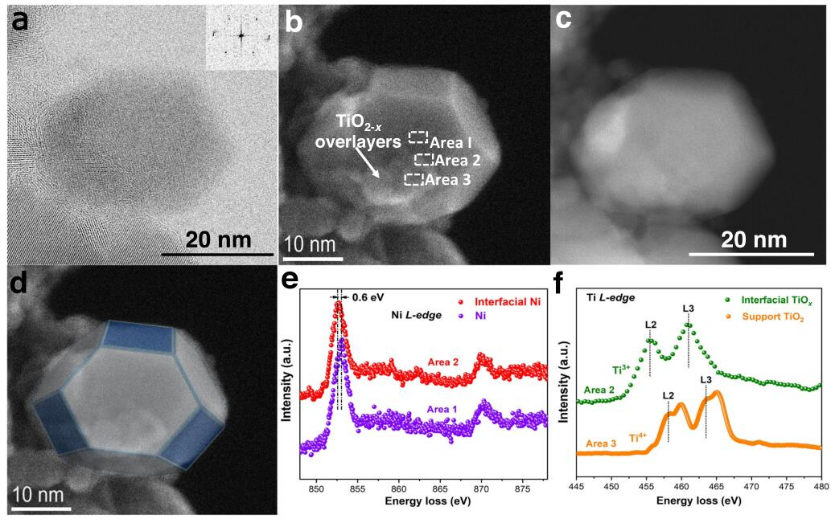
圖3 TiO2-x/Ni-450催化劑的結構和形貌。(a)STEM-BF(b)STEM-SE(c)TiO2-x/Ni-450催化劑在H2氣氛(10Pa)中450°C還原后的結構和形貌;(d)鎳顆粒示意圖。分別在Spot I和II收集的(e)Ni L-邊原位電子能量損失譜(EELS);分別在Spot I和II收集的(f)Ti L-邊光譜。
鈍化的TiO2-x/Ni-450催化劑在H2氣氛中(壓力:10Pa)在450℃下被還原1h,通過FFT圖案(圖3a插圖)確定還原的金屬鎳納米粒子的FCC微觀結構。圖3b顯示,在450℃下鈍化TiO2-x/Ni-450后,具有良好有序形狀的還原鎳顆粒被TiO2-x覆蓋了一部分。圖3d說明了Ni NP的表面結晶平面由低指數的結晶平面終止。為了證明金屬納米顆粒和氧化物之間的特定相互作用,在I區(鎳顆粒)、II區(界面)和III區(TiO2支撐體)獲得了EELS鎳L邊和TiL邊譜,以便在圖3e,f中區分Ni NP的主體、界面Ni原子和Ti原子以及TiO2支撐體的Ti原子的電子特性。與金屬顆粒表面的Ni物種(區域I)相比,界面(區域II)上的Ni物種的L-邊朝向低能量偏移(0.6 eV),表明Niδ-物種的存在。在界面上可以觀察到豐富的Ti3+物種,而TiO2載體上(區域III)則沒有。
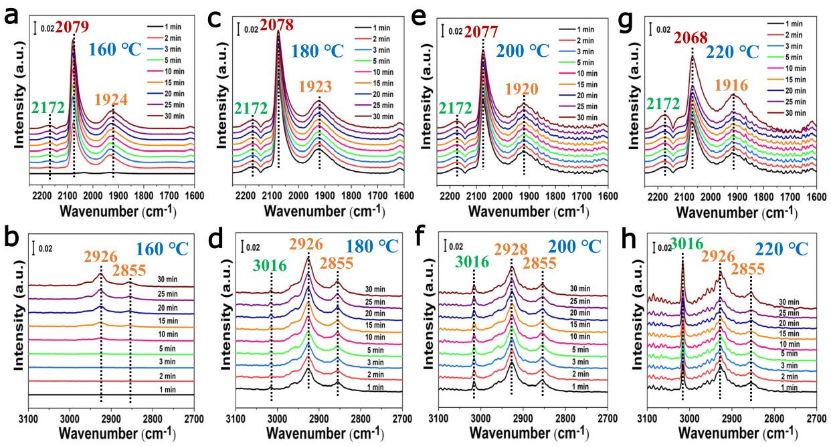
圖4CO加氫反應機理的原位紅外光譜研究。分別在(a,b)160℃,(c,d)180℃,(e,f)200℃,(g,h)220℃下對TiO2-x/Ni-450催化劑進行的原位時間分辨DRIFTS譜測試。
使用原位時間分辨DRIFTS來監測在不同件下TiO2-x/Ni-450催化劑上進行CO氫化過程中反應分子的動態演變(圖4)。在160℃的反應條件下,觀察到~2079 cm-1和~1924 cm-1的主要紅外帶,分別為Ni缺陷位點上的線性吸附CO物種和Niδ-位點上的橋式吸附物種(圖4a)。在~2926 cm-1和~2855 cm-1的紅外帶,為CH2*物種的不對稱vas(C-H)和對稱vs(C-H)拉伸振動,隨著時間延長強度逐漸增加(圖4b),表明C2+烷烴產物的形成。對于催化CO氫化的TiO2-x/Ni-450來說,在~3016 cm-1處(CH4分子的v(C-H)拉伸振動)沒有發現明顯的紅外帶。這表明,在160℃時,C2+烴類產物是CO加氫的主要產物,而不是CH4。當反應溫度上升到180℃時,在~2079 cm-1和~1924 cm-1的紅外譜帶不發生明顯變化(圖4c)。在~3016 cm-1處的一個非常弱的紅外帶是由CH4分子的C-H拉伸模式引起的。如圖4d所示,~2926 cm-1和~2855 cm-1處的紅外帶強度增加,表明隨著FTS的催化性能提高,更多的C2+烷烴形成。隨著反應溫度依次升高到220℃,~2068 cm-1和~1916 cm-1的紅外帶明顯減少,這是因為催化性能的大幅提高(圖4g)。在圖4h中,CH4分子在~3016 cm-1處的C-H拉伸模式大幅增加,同時在~2926 cm-1和~2855 cm-1處出現了大量的CH2*物種,表明通過FTS對甲烷化和C2+烷烴產品的催化活性同時增強。
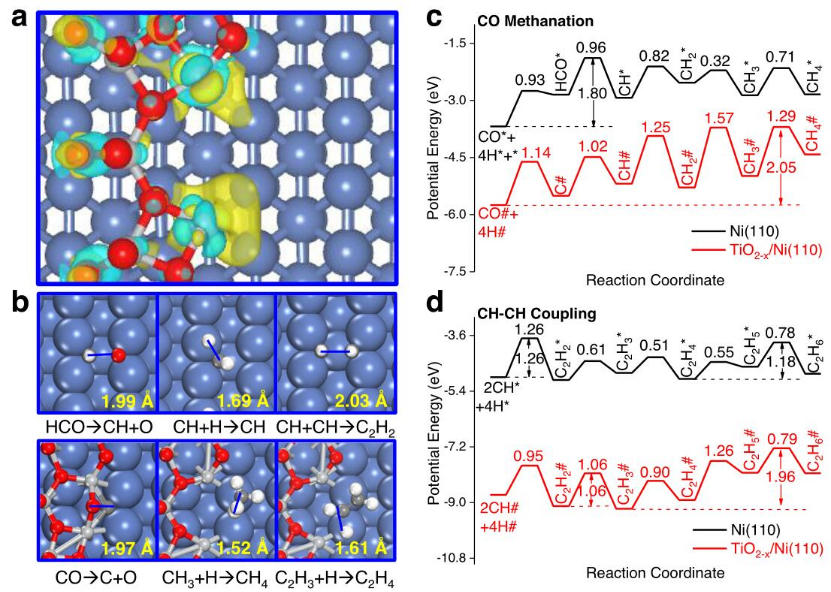
圖5DFT研究反應機理。
采用DFT計算研究TiO2-x/Ni(110)催化劑上的CO氫化機制。構造了Ti6O11/Ni(110)表面,即在Ni(110)表面沉積一層保持TiO2(101)表面特征的Ti6O11簇,來模擬界面TiO2-x/Ni(110)催化劑。Ti6O11團簇中的所有Ti原子和8個O原子都傾向于與Ni(110)表面結合,使得Ti6O11團簇和Ni(110)表面之間發生大量電荷轉移(圖5a)。CO氫化是首先通過CO活化和氫化生成CHx(x=0-3)單體,這些單體可以進一步氫化為甲烷(CH4)和/或相互耦合形成C2+烷烴/烯烴(圖5b)。Ti6O11/Ni(110)和Ni(110)表面表現出不同的CO活化和進一步氫化機制。CH加氫到CH4在Ni(110)表面的最高活化壘為0.82eV。然而,通過CH-CH、CH-CH2和CH2-CH2耦合形成的C2H6在Ni(110)表面上具有較高的活化屏障1.21-1.26 eV(圖5c,d)。因此,Ni基表面上CO氫化的主導產物CH4而不是C2H6。
與Ni(110)表面不同,在Ti6O11/Ni(110)表面上,直接的CO解離路徑比通過甲酰(HCO)路線單體的氫輔助CO活化更容易。因此,Ti6O11/Ni(110)表面比Ni(110)表面的活性高得多。Ti6O11/Ni(110)對CO活化的更高活性可以歸因于Ti原子和O原子之間的強結合力降低了相應的過渡態能量和CO直接解離的反應能量。在Ti6O11/Ni(110)的Niδ-/TiO2-x界面上,強烈吸附的解離的C原子向CH4的氫化是吸熱反應,因此導致CH4形成的活性很低,活化能壘很高(圖5c)。相比之下,在Ti6O11/Ni(110)表面的Niδ-/TiO2-x界面上,通過CH-CH耦合步驟形成的C2H4中間物比CH4產物的生成要有利得多,其活化能壘為1.06 eV(圖5d)。
05、總結與展望
本文通過調節強金屬-載體相互作用(SMSI)有效調節了界面催化劑的幾何和電子結構。電子顯微鏡、多種原位光譜表征和密度泛函理論計算的綜合研究表明,TiO2-x/Ni催化劑中Niδ-/TiO2-x界面位點的存在,可以強烈結合碳原子,抑制甲烷的形成,并促進C-C鏈的耦合,使得C2+碳氫化合物在Ni表面的產生。本工作不僅發現了一種具有獨特界面結構的新型SMSI催化劑用于大氣壓下的合成氣轉化,而且深入了解了SMSI效應所驅動的界面協同催化作用。
審核編輯:郭婷
-
數據
+關注
關注
8文章
7067瀏覽量
89131 -
納米
+關注
關注
2文章
697瀏覽量
37025
原文標題:Nature Commun.:TiO2?x/Ni界面促進費托合成制備高碳烴
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
如何提高濕法刻蝕的選擇比
選擇性沉積技術介紹
V2G技術在智能電網中的作用
SMT工藝分享:0.4mm間距CSP
微小無鉛釬焊接頭中金錫化合物的形貌與分布:激光與熱風重熔方法的比較
過電流保護的選擇性是靠什么來實現的
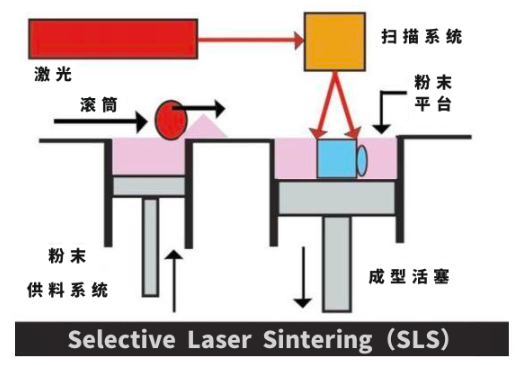
物聯網行業中3D打印工藝——SLS(選擇性激光燒結)工藝
FLIR光學氣體成像熱像儀的高靈敏度模式
功率合成電路的主要技術要求是什么
交流二元繼電器如何具有相位選擇性和頻率選擇性
制冷劑的發展歷程與發展趨勢
鍺化硅(SiGe)和硅(Si)之間的各向同性和選擇性蝕刻機制





 工商網監
工商網監
評論