


01
導讀
眾所周知,陰極氧還原反應(ORR)涉及多電子轉移步驟,使得反應動力學十分緩慢,進一步限制了質子交換膜燃料電池等清潔能源轉化裝置的輸出功率。目前,Pt仍然是廣泛應用的商業電催化劑之一,但因其使用成本較高、儲量稀缺而無法滿足日益增長的商業需求。Fe-N-C催化劑因其具有高的本征ORR活性,而被認為是最具有應用前景的非貴金屬催化劑之一。然而,在酸性介質下,Fe-N-C催化劑的ORR活性以及耐久性仍然不能達到實用指標。
另一方面,近年來,雙原子催化劑的發展如火如荼。以Fe-N-C催化劑為基礎,近年來已逐漸開發出了Fe-Mn、Fe-Co、Fe-Ni等雙原子催化劑,在相關的電催化體系也顯示出了優于單原子催化劑的電化學性能。與后過渡金屬相比,將前過渡金屬引入Fe-N-C的研究報道相對較少。前過渡金屬具有的更多的未占據d軌道以及親氧特性,有望地進一步調節Fe中心的ORR活性。
02
成果背景
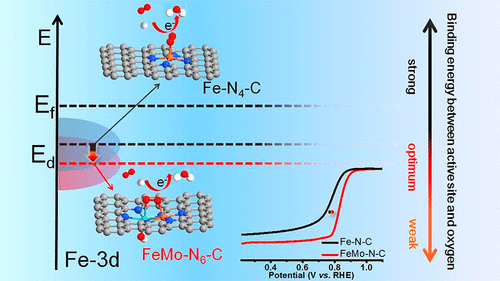
首都師范大學孫文明,清華大學王定勝、李亞棟院士等人成功在N摻雜碳基底上引入了Fe-Mo雙原子位點,并記為FeMo-N-C。研究發現,引入的Fe-Mo原子對可以有效地調控Fe的電子構型,并使Fe的d帶中心發生負移,從而削弱對ORR中間體的吸附,最終促進ORR。
因此,FeMo-N-C在酸性電解質中表現出良好的ORR活性,起始電位高達0.98 V,半波電位達0.84 V。相關成果以《Regulating the FeN4Moiety by Constructing Fe–Mo Dual-Metal Atom Sites for Efficient Electrochemical Oxygen Reduction》為題發表于國際頂尖雜志《NanoLetters》上。
03 關鍵創新
(1)本文首次設計并制備了一種新型的Fe-Mo雙原子催化劑,經過詳細的結構表征,證實了Fe、Mo主要以FeMo-N6的結構形式高度分散于碳載體上;
(2)在酸性介質下,FeMo-N-C表現出優異的ORR性能,其半波電位達到了0.84 V,遠優于Fe-N-C以及其他文獻所報道的非貴金屬催化劑的性能;
(3)理論計算揭示了在新型的FeMo-N6位點中,引入的Mo可以調節Fe中心的電子構型,使d帶中心負移,從而削弱了對ORR中間體的吸附,加速了ORR。
04
核心內容解讀
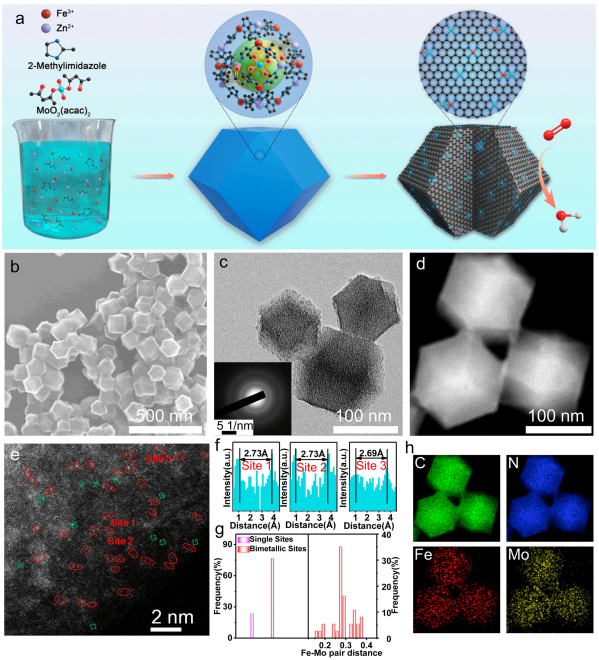
圖1合成示意圖及結構表征:(a)FeMo-N-C的合成示意圖;(b) FeMo-N-C的SEM圖像;(c) TEM圖像和相應的選取電子衍射圖案;(d) HAADF-STEM圖像;(e)像差校正HAADF-STEM圖像;(f,g)對圖e的選定區域進行原子強度分析以及Fe-Mo雙位點的占比、原子間距進行統計;(h)圖d相應的EDS映射。
本文采用主客體策略合成了含有Fe-Mo雙原子位點的FeMo-N-C催化劑。如圖1a所示,乙酰丙酮鉬被封裝在原位生長的FeZn-ZIF骨架中,將所得的Mo-FeZn-ZIF進行熱解可得多孔碳基催化劑。圖1b、c的SEM、TEM圖像顯示,FeMo-N-C仍然保留了十二面體形貌,直徑約100 nm。
相應的選區電子衍射(SAED)圖案表明該多孔碳的結晶度相對較高,有利于增強導電性。圖1d的HAADF-STEM圖像顯示該多孔碳上未存在明顯的納米顆粒。采用像差校正的HAADF-STEM圖像對金屬原子的分散形式進行辨別,如圖1e所示,可以看到碳基底上分布大量的Fe/Mo雙原子對(紅圈標識),其對應的電子能量損失譜(EELS)表明這些原子對由Fe和Mo所構成。
另一方面,少量單個Fe或Mo原子用綠圈標出。對圖1e中標記的區域1、2和3的Fe-Mo雙金屬位點距離進行測量,如圖1f所示,其平均間距約為0.27 nm。圖1g的統計結果顯示,大部分(~70%)亮點的原子間距為0.3 nm,也說明了催化劑中具有大量的Fe-Mo雙原子位點。通過相應的EDS映射(圖1h),進一步驗證了Fe、Mo和N元素在碳載體中均勻分布。
最后,ICP-MS測試結果顯示Fe和Mo的負載量分別為0.78和0.71 wt %。
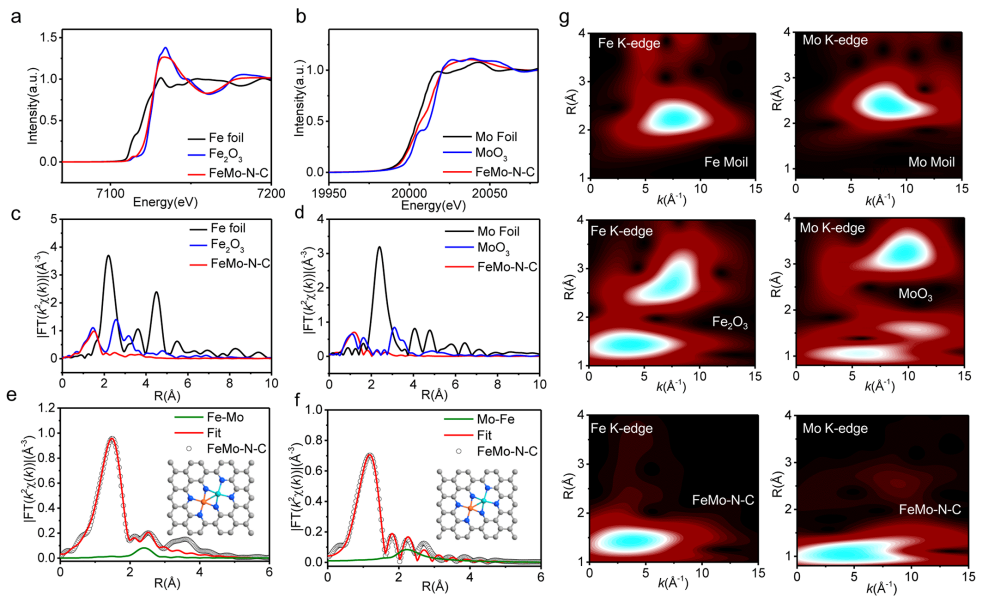
圖2基于XAFS的結構分析:(a) Fe的K邊XANES譜圖;(b) Mo的K邊XANES譜圖;(c,e) Fe的K邊FT-EXAFS譜圖及其擬合結果;(d,f) Mo的K邊FT-EXAFS譜圖及其擬合結果;(g)小波變換。
采用XAFS對FeMo-N-C催化劑的原子精細結構進行解析。如圖2a的Fe的K邊XANES譜圖所示,FeMo-N-C的近邊吸收譜線介于Fe與Fe2O3之間,說明其Fe物種處于正價態(+0<δ<+3)。類似地,在Mo的K邊XANES譜圖中,FeMo-N-C的近邊吸收譜線介于Mo與MoO3之間,即Mo物種處于正價態(+0<δ<+6)。
圖2c、d的Fe、Mo的K邊FT-EXAFS譜圖顯示,FeMo-N-C分別在~1.5 ?、1.2 ?處出現主峰,分別對應Fe-N、Mo-N配位,同時均在2.2 ?、2.4 ?處未發現明顯的Fe-Fe、Mo-Mo配位峰,從而支持了電鏡觀察結果。對譜圖進行擬合,結果如圖2e、f所示,結果顯示說明Fe-N、Mo-N的配位數均為4,且譜圖均在~2.7 ?處出現了弱配位峰,結合上述統計結果,可以將其歸為Fe-Mo配位峰。
圖2g的小波變換顯示,FeMo-N-C中未發現Fe-Fe與Mo-Mo信號,進一步支持了這一結果。因此,Fe、Mo以FeMo-N6的結構形式分散于碳載體上。
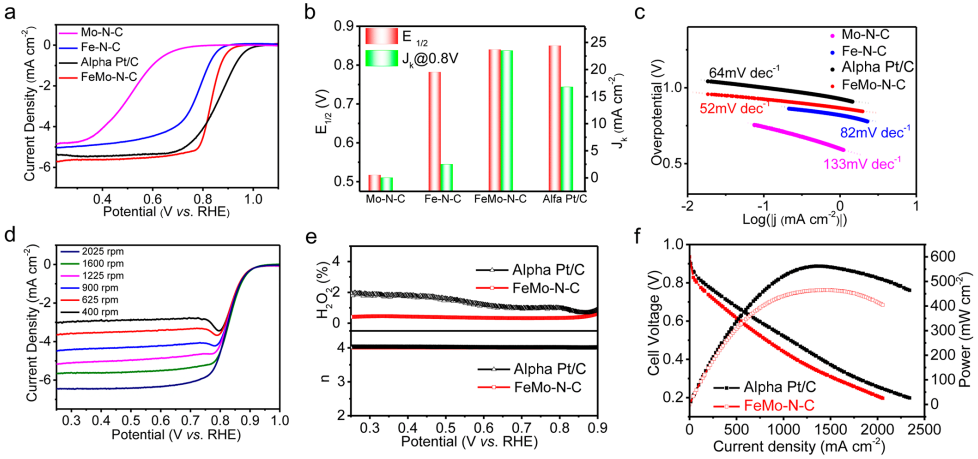
圖3 電催化ORR性能:(a)在O2飽和的0.1 M HClO4溶液下的LSV曲線;(b)半波電位以及在0.8 V下的動力學電流密度比較;(c)不同催化劑的Tafel斜率比較;(d) FeMo-N-C在不同轉速下的極化曲線;(e)基于RRDE測試得到的轉移電子數與H2O2產量;(f)基于FeMo-N-C的H2-O2燃料電池的放電極化曲線與放電功率密度。
采用標準的三電極測試體系研究了催化劑的ORR性能。在含飽和O2的0.1M HClO4溶液中,圖3a顯示了各催化劑的極化曲線。其中,FeMo-N-C顯示出了優異的ORR性能,其起始電位高達0.98 V,同時半波電位達0.84 V,優于Fe-N-C、Mo-N-C,同時也是目前報道的最佳的非貴金屬基酸性ORR電催化劑之一。
同時,如圖3b所示,在0.8 V下FeMo-N-C的動力學電流密度為23.5 mA cm-2,遠大于Fe-N-C和Mo-N-C的動力學電流密度之和,說明Fe-N-C和Mo-N-C位點之間存在協同作用。由極化曲線衍生的Tafel曲線(圖3c)顯示,FeMo-N-C表現出低至52 mV dec-1的Tafel斜率,具有最快的ORR動力學。
對不同轉速下的極化曲線進行K-L方程擬合,得到FeMo-N-C的轉移電子數為3.99。同時,圖3e的RRDE測試顯示n接近4,H2O2產率低于1.0%。兩種測試共同說明了FeMo-N-C遵循了高效的四電子反應機制。
最后,將FeMo-N-C作為陰極催化劑,并用于H2-O2燃料電池中,如圖3f所示,該電池的峰值功率密度為460 mW cm-2,約為Pt/C基燃料電池的83%,并超過了大多數非貴金屬催化劑所報道的輸出性能。
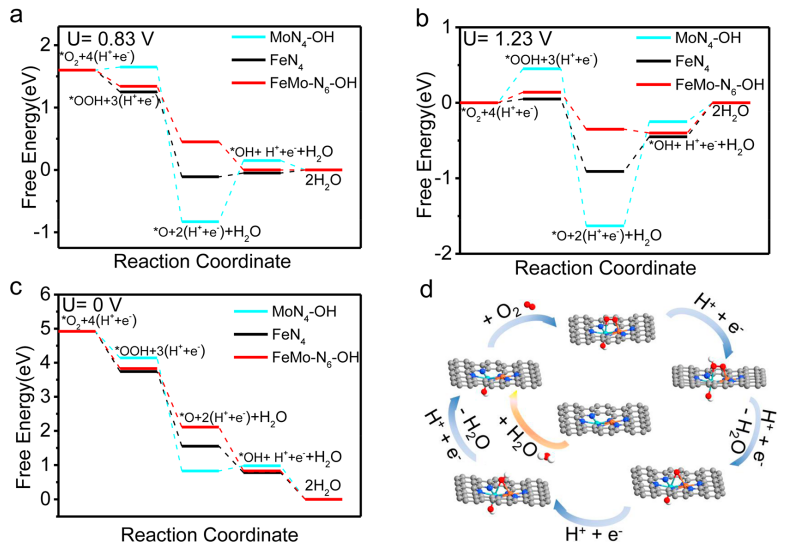
圖4催化機理分析:(a-c)在不同外加電位下,FeMo-N-C、Fe-N-C與Mo-N-C的ORR自由能分布;(d) FeMo-N-C的ORR機制。
采用DFT計算研究了FeMo-N-C的ORR機制。首先,在整個工作電位下,*OH在FeMo-N-C上的吸附是熱力學自發的,因此形成了FeMo-N6-(OH)構型。類似的結果也存在于MoN4位點上。相比之下,FeN4位點上不存在軸向OH配體吸附。如圖4a所示,在U=0.83 V,FeMo-N6-(OH)位點在整個ORR過程中均是熱力學有利的,而MoN4-(OH)位點與FeN4位點在*O向*OH的轉化步驟均為熱力學吸熱過程。
另外,根據圖4b、c的計算結果,可以看出在整個工作電位下,MoN4-(OH)位點上均存在吸熱步驟,導致其ORR活性較差;而FeN4位點雖然表現出更有利的O2質子化過程,但對于后續反應中間體的吸附過強,導致其ORR活性低于FeMo-N6-(OH)位點。因此,理論計算分析驗證了這種新型的FeMo-N6-(OH)位點的優勢,從而極大地促進了ORR性能,其反應機制如圖4d所示。
05
成果啟示
本文設計了一種新型的雙原子Fe-Mo電催化劑,并在酸性電解質下表現出超強的ORR活性。由于O2更傾向于以橋鍵形式吸附于Fe-Mo原子對上,O-O鍵更容易發生斷裂,從而有利于促進ORR。另一方面,引入的Mo可以調節Fe中心的電子構型,使d帶中心負移,從而削弱了對ORR中間體的吸附,加速了ORR。因此,FeMo-N-C在半電池與全電池的測試過程均顯示出了優異的性能。總之,該工作為雙原子催化劑的設計提供了新的見解,加速了非貴金屬催化劑在燃料電池的應用。
審核編輯:劉清
-
電解質
+關注
關注
6文章
821瀏覽量
20812 -
EDS
+關注
關注
0文章
100瀏覽量
11945
原文標題:孫文明/王定勝/李亞棟Nano Letters:雙原子Fe-Mo位點,超強酸性ORR活性
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
酸性溶液清洗劑的濃度是多少合適

隆基重磅發布Hi-MO X10輕質雙防組件
DAC5672/62/52 14位和12位雙通道DAC EVM用戶指南

原子結構模型及特點 原子的組成及結構解析
隆基Hi-MO 9及BC技術客戶推介會成功舉辦
隆基推出全新Hi-MO 9和Hi-MO X10組件
新品速遞 | 麥科信(Micsig)推出MO 3系列高分辨率模塊化示波器
MO管在射頻放大器中的使用
中國科大在納米級空間分辨紅外成像及催化研究中取得新進展
Wolfspeed助力捷豹TCS車隊征戰FE電動方程式上海站
PP強酸強堿氮氣柜和普通氮氣柜的區別及共同點

探索 32KHz MEMS 振蕩器的卓越性能:μPower MO1532、MO1552、MO1630、MO1566 和 MO1568

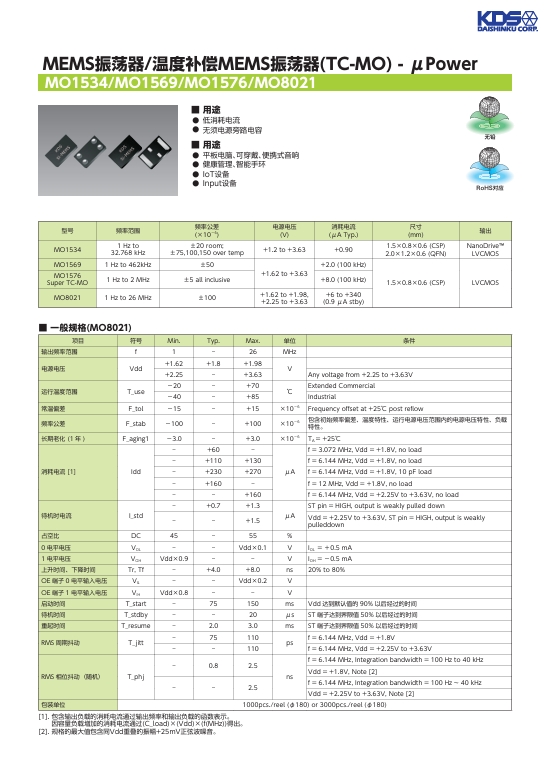
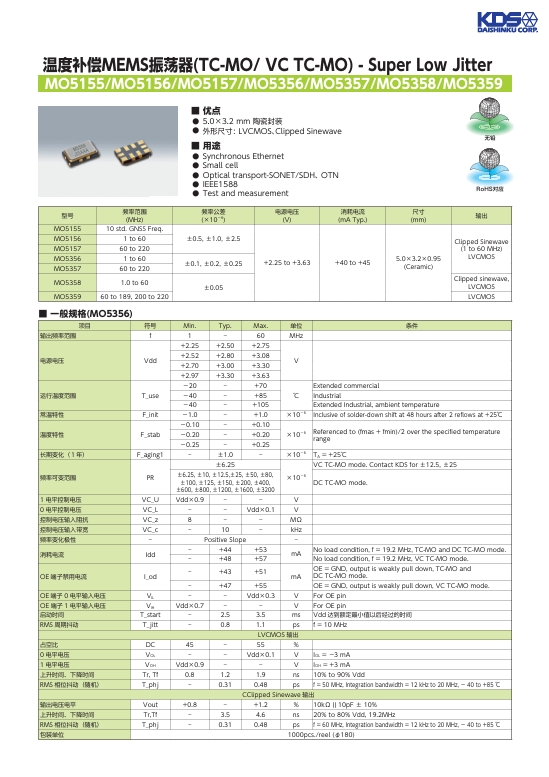
深入了解 MEMS 振蕩器 溫度補償 MEMS 振蕩器 TC-MO
溫度補償 MEMS 振蕩器 TC-MO/VC TC-MO
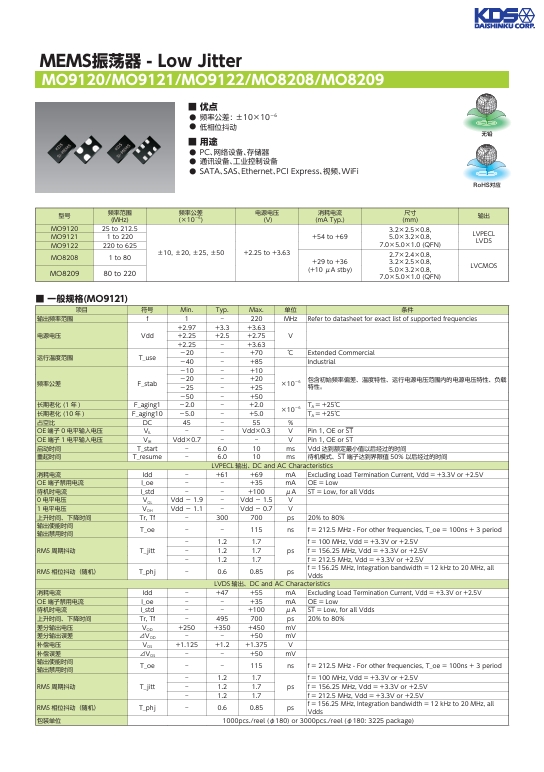
MEMS 振蕩器:Low Jitter MO9120 MO9121 MO9122 MO8208 MO8209 的卓越性能與應用





 工商網監
工商網監

評論