 單層NiFeB氫氧化物納米片中的Ni氧化態轉變助力高效OER
單層NiFeB氫氧化物納米片中的Ni氧化態轉變助力高效OER
研究背景
高氧化態的過渡金屬位點(如NiOOH中氧化態大于+3的Ni位點),被認為是析氧反應(OER)的活性位點。通過形成高氧化態的過渡金屬位點,可以降低電催化反應的起始電位,從而加快OER的動力學。迄今為止,人們致力于開發高效的鎳基催化劑。
然而,通過人為提高Ni的氧化態來提高OER活性,相關研究卻相當有限。當鎳基催化劑與等離子體納米顆粒偶聯時,在可見光照射下,納米顆粒中產生了帶正電荷的“空穴”,這很容易促進附近的Ni2+氧化成高氧化態的物質,獲得更高的OER活性。
Ni基氧化物催化劑與其他金屬氧化物(如MoO2和WOx)和非金屬(如磷酸鹽)偶聯,也能有效形成OER所需的高氧化態活性位點。因此,使用這些催化劑可以大幅度提高OER活性。盡管在先前的報道中已取得不錯的成果,但OER活性仍遠未達到最佳狀態。進一步探索促進Ni氧化的有效策略,將OER活性提高至前所未有的水平,仍然是非常迫切的需求。
研究表明,在傳統的NiFe氫氧化物催化劑中引入缺電子硼(B),可以有效地促進Ni的氧化態轉變,顯著提高OER活性。通常,直接從Ni2+中提取電子以形成所需的Ni3+δ是一個困難的過程,只能在高電位下實現。預計在Ni附近的B可能作為參與氧化過程的電子流中轉站,參與到Ni氧化過程中。由于其固有的缺電子特性, 轉運B位可能作為一個“電子穴”,促進電子從Ni2+位流動,從而允許在較低的電位下形成活性Ni3+δ,從而提高OER活性。
成果介紹
為了驗證這一想法,西安交通大學高傳博教授和蘇州大學程濤教授通過原位水解NiFeB合金納米粒子合成了單層NiFeB納米片。拉曼光譜、X射線吸收光譜和電化學測量結果表明,NiFeB氫氧化物納米片中Ni2+(OH)2轉化為Ni3+δOOH的電位低于無B的NiFe氫氧化物納米片。
密度泛函理論(DFT)計算表明,Ni原子在加入B后呈現較高的氧化態,表明Ni和B組分之間存在電子相互作用。結果表明,NiFeB氫氧化物納米片的OER活性達到100 mA cm?2,過電位為252 mV,優于無B的NiFe氫氧化物納米片的OER活性(過電位為337 mV)和目前報道的大多數鎳基催化劑。
雖然硼酸鹽已被用作電解質添加劑,過渡金屬硼化物已被直接用作OER的催化劑,但B的作用尚不清楚。這項工作揭示了B的缺電子特性在促進Ni氧化態轉變方面的作用,為設計先進的金屬氫氧化物(或氧化物)催化劑提供了新的機會,提高了電化學裂解水在應用上的可行性。
圖文介紹
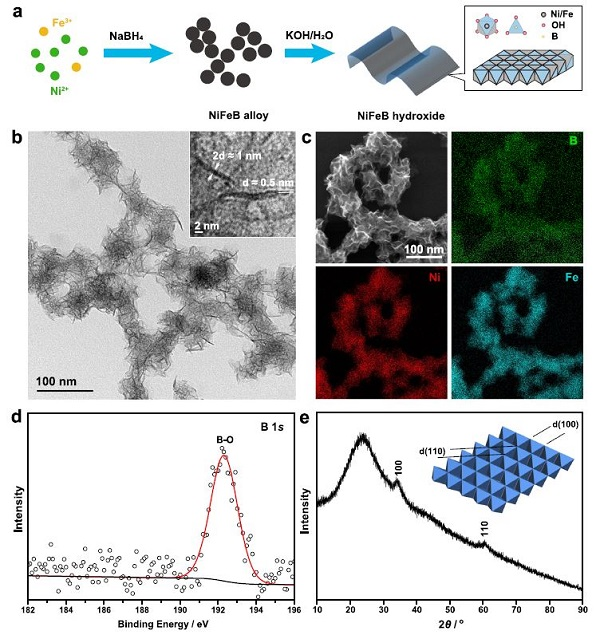
圖1.單層NiFeB氫氧化物納米片的合成與表征。(a)單層NiFeB氫氧化物納米片的合成方法。(b)單層NiFeB氫氧化物納米片的TEM圖像。(c)Ni, Fe, B的EDS元素圖。(d)B 1sXPS。(e)單層NiFeB氫氧化物納米片的XRD。
在環境條件下,通過NiFeB合金納米粒子原位水解,合成單層NiFeB氫氧化物納米片(圖1a)。首先,在超聲作用下,用NaBH4在乙醇溶液中還原Ni(NO3)2和Fe(NO3)3,合成了NiFeB合金納米顆粒。NaBH4既是還原劑又是硼源。接下來,將NiFeB合金納米顆粒分散在KOH溶液中,將其轉化為氫氧化物。
NiFeB合金納米顆粒經過快速水解,演變為超薄納米片(圖1b)。這些納米片被纏繞成大量褶皺的束,測量到的皺紋厚度約為1 nm,相當于2層納米片(圖1b,附圖)。因此,單層納米片的厚度約為0.5 nm。能量色散X射線能譜(EDS)元素映射圖顯示,Ni、Fe和B在納米片中均勻分布,形成均勻的固溶體,而不是分離相(圖1c)。
XPS顯示,水解后零價產物消失,僅顯示氧化產物(Ni2+、Fe3+和B-O)的信號;這表明納米片是由NiFeB氫氧化物組成的,即通過橋接O或OH連接的陽離子Ni、Fe、B(圖1d)。XRD顯示了位于~34°和60°的衍射峰,分別來自金屬氫氧化物中MO6(M = Ni, Fe)八面體的(100)和(110)晶面,與NiFe-LDHs中觀察到的結構相似(圖1e)。
沒有觀察到其他衍射峰,特別是來自共邊MO6八面體的反射峰,這表明納米片是由單一的MO6層組成的。
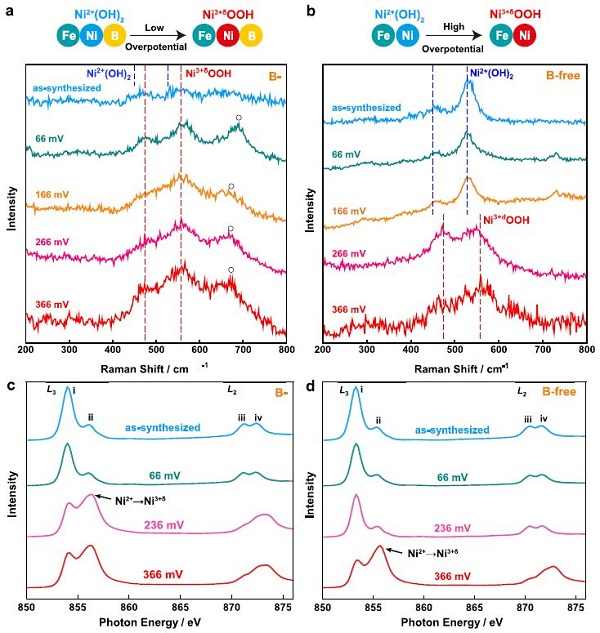
圖2.NiFeB氫氧化物在不同過電位下的(a,b)拉曼光譜;(c,d)Ni L3,2邊XAS光譜。
為了揭示B的作用,作者收集了NiFeB氫氧化物納米片的拉曼光譜,以監測Ni在不同過電位下的氧化態轉變(圖2a)。NiFeB氫氧化物納米片的光譜中沒有明顯的拉曼信號。當過電位增加到66mV時,在~474和558 cm-1處出現了清晰的拉曼峰,對應于Ni3+δOOH。
當過電位增加到166、266和366mV時,這些峰值保持不變。位于680 cm-1處的寬拉曼峰可以歸因于NiFeB氫氧化物納米片中的Fe-O。實驗結果表明,在所有過電位下,NiFeB氫氧化物納米片中的Ni均以Ni3+δOOH的形式存在。NiFeB氫氧化物納米片在過電位為66mV或更高時形成了具有電催化活性的Ni3+δOOH。
作者研究了超薄無B的NiFe氫氧化物納米片在不同過電位下的拉曼光譜(圖2b)。NiFe氫氧化物納米片在449和527 cm-1處有明顯的拉曼峰,這分別是由于Ni2+(OH)2中Ni2+ -OH和Ni2+-O鍵的振動引起的。
這些峰在過電位為0 ~ 166mV的范圍內保持不變,表明在此條件下納米片中的Ni保持為Ni2+(OH)2,其OER活性不高。只有當過電位增加到266mV或更高時,才會出現OER的活性Ni,即Ni3+δOOH。因此,在無B的NiFe氫氧化物納米片中,形成有OER活性的Ni3+δOOH是一個更為困難的過程。
作者進一步利用XAS驗證了NiFeB和NiFe氫氧化物納米片中Ni在不同過電位下的氧化態轉變(圖2c, d)。測試前,納米片保持在不同的過電位(66,236和366mV)。在Ni L3,2邊XAS譜中,i和iii峰是由八面體晶體場中Ni 2p軌道上的電子躍遷到3dt2g軌道上產生的,ii和iv峰是由Ni 2p軌道上的電子躍遷到3deg軌道上產生的。
ii/i峰強度比反映了eg和t2g軌道上的空穴比例,是衡量催化劑中Ni氧化態的指標。這些結果證實了B在促進Ni氧化態轉變中的關鍵作用,與拉曼光譜顯示的趨勢一致。
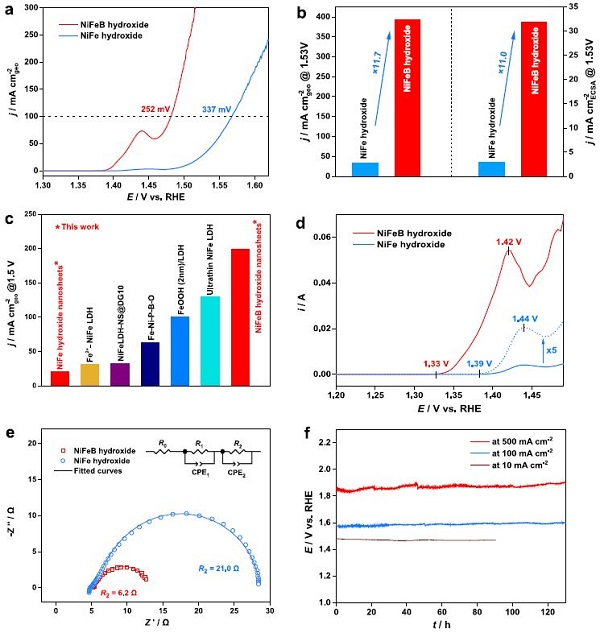
圖3.電催化析氧性能比較。(a)OER極化曲線 (95% iR補償)。(b) 不同催化劑在1.53 V下觀察到的電流密度比較。(c) NiFeB氫氧化物納米片與NiFe氫氧化物納米片及典型非貴金屬催化劑的OER活性比較。(d) 差分脈沖伏安曲線 (95% iR補償)。(e)在恒定電位為1.50 V vs. RHE時的奈奎斯特圖。(f) 催化劑在電流密度為10、100和500 mA cmgeo?2下的時間電位曲線。
用線性掃描伏安法在O2飽和的1.0M KOH中測量OER活性(圖3a)。NiFeB氫氧化物納米片的催化活性明顯高于無B的NiFe氫氧化物納米片。為了達到100mA cmgeo-2的電流密度,NiFeB氫氧化物納米片需要比NiFe氫氧化物納米片(337mV)低得多的過電位(252mV)。NiFeB氫氧化物納米片在1.53 V vs. RHE條件下,即過電位為300mV時,電流密度為393mA cmgeo-2,是NiFe氫氧化物納米片(33.6mA cmgeo-2)的11.7倍(圖3b)。
就1.5 Vvs RHE下的電流密度而言,與文獻報道的典型非金屬催化劑相比,NiFeB氫氧化物是OER的首選催化劑(圖3c)。通過差分脈沖伏安法(DPV)研究Ni氧化峰,進一步研究了Ni2+(OH)2→Ni3+δOOH躍遷的電化學過程(圖3d)。NiFeB和NiFe納米片的氧化峰起始電位分別為1.33和1.39 V。
因此,將B摻入到NiFe氫氧化物催化劑中,引起了Ni氧化峰的60 mV負移。B的峰位也發生了20mV的負移。這一結果再次強調了B在低過電位下促進高氧化態Ni形成的作用,與Raman和XAS觀察結果一致。值得注意的是,NiFeB氫氧化物納米片的Ni氧化峰明顯高于NiFe氫氧化物納米片的Ni氧化峰,表明在B的促進下,NiFeB氫氧化物催化劑形成了更多的活性Ni3+δOOH物種。NiFeB氫氧化物催化劑具有較低的起始電位和較多的活性Ni3+δ物種,可能是其具有優異OER活性的原因。
為了更好地評價NiFeB氫氧化物納米片的本征活性,作者檢測了催化劑對ECSA歸一化的OER電流密度(圖3b)。LSV數據表明,NiFeB氫氧化物納米片的性能優于無B的NiFe氫氧化物納米片。在1.53 V下,NiFeB氫氧化物納米片的電流密度為31.8mA cmECSA-2,是NiFe氫氧化物納米片(2.9mAcmECSA-2)的11.0倍。
上述結果證實了B在提高NiFeB氫氧化物催化劑OER本征催化活性方面起到關鍵作用。圖3e顯示了催化劑在1.50 V下的Nyquist圖。NiFe和NiFeB氫氧化物納米片的電化學電荷轉移電阻分別為21.0和6.2 Ω。NiFeB氫氧化物的電荷轉移阻抗比NiFe氫氧化物低得多,這與NiFe催化劑的OER活性非常吻合。
單層NiFeB氫氧化物納米片在OER中也表現出較高的穩定性。圖3f顯示了NiFeB氫氧化物納米片在恒定電流密度為10、100和500 mA cm?2時的時間電位曲線。NiFeB氫氧化物納米片維持電流密度所需的電勢在130 h內保持穩定。
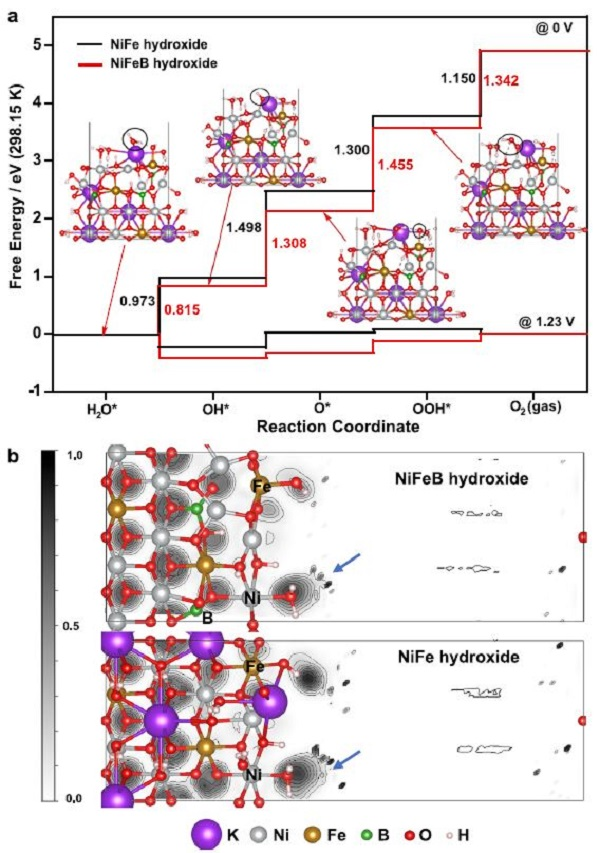
圖4. DFT 計算。(a) NiFeB和NiFe氫氧化物電催化OER的自由能圖。(b)NiFeB(上)和NiFe氫氧化物(下)的電子局域函數(ELFs)。
根據DFT計算,B的引入降低了OER的能壘。通過研究OER中間體的自由能來預測NiFeB氫氧化物和NiFe氫氧化物催化劑的OER活性(圖4a)。考慮了由*H2O到O2的反應路徑上有*OH,*O和*OOH中間體的四步反應路徑。計算了兩種電位(0和1.23 V)下的自由能,結果顯示自由能沿反應坐標的變化趨勢相似。
0 V時,NiFe氫氧化物形成*O (*OH→*O)為電位決定步驟(PDS),自由能差為1.498 eV,與前人結果一致。引入B后,NiFeB氫氧根上*O的生成能降至1.308 eV。而*OOH (*O→*OOH)的生成能增加到1.455 eV,*OOH形成了PDS。NiFeB氫氧化物的OER活性提高,說明了B在提高NiFeB氫氧化物催化劑OER活性中的關鍵作用。
DFT計算還表明,在NiFe氫氧化物中引入B后,Ni的氧化態升高。這些電子變化由ELFs表示(圖4b)。采用灰度格式來闡明ELFs,其中0.0(白色)表示完全離域狀態,1.0(黑色)表示完全局域狀態。引入B后,Ni結合OH(如圖中箭頭所示)的電子局域化增強了,這為B附近Ni的氧化態增加提供了間接證據。
為了定量評估這些電荷,進行了Bader電荷分析,結果顯示形式電荷最大值為+0.06。電子相互作用使B對Ni元素施加電子下沉效應,促進氧化態轉變,有效降低了OER的起始電位,因此具有良好的催化活性。
總結
作者通過將NiFeB合金納米粒子在堿性介質中水解,合成了單層NiFeB氫氧化物納米片。TEM、XRD和XPS結果證實了MO6的單層形成(厚度約0.5 nm),未發生層間堆積。NiFeB氫氧化物納米片在堿性電解質中表現出優異的催化活性和顯著的長期穩定性。
只需要252mV的過電位就可以達到100mA cmgeo?2的電流密度,優于文獻報道的大多數非貴金屬OER催化劑。拉曼光譜、XAS和電化學DPV分析表明,OER活性與Ni2+向Ni3+δ的轉變有關,在NiFe氫氧化物中加入B可有效降低Ni3+δ的電位。
更具體地說,DPV分析表明,將B加入到NiFe氫氧化物催化劑中,導致了啟動Ni氧化態轉變的電位發生了60 mV的負移。DFT結果表明,B增加了Ni的氧化態,改變了OER的速率決定步驟,降低了能壘,解釋了實驗結果。所有這些結果都支持了預想的假設,即B促進高氧化態Ni3+δ形成,生成OER所需的活性物種。
結果表明:(1)將B加入NiFe氫氧化物是制備高效OER催化劑的有效途徑;(2) B的加入降低了Ni2+氧化為活性Ni3+δ基團所需的電位,降低了OER的起始電位,增強了OER的動力學;(3)在電催化OER過程中,缺電子的B可能起到了促進Ni2+氧化為Ni3+δ的作用。此研究為鎳基催化劑的電荷工程提供了一個穩健的策略,在高性能電化學水裂解應用中實現高效的OER。
文獻鏈接:
https://doi.org/10.1038/s41467-022-33846-0
審核編輯:劉清
-
電解質
+關注
關注
6文章
814瀏覽量
20084 -
XRD
+關注
關注
0文章
133瀏覽量
9091 -
EDS
+關注
關注
0文章
96瀏覽量
11537
原文標題:Nature子刊:單層NiFeB氫氧化物納米片中的Ni氧化態轉變助力高效OER
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
金屬氧化物和柔性石墨烯MOS的區別

多晶氧化物中的晶界和異質界面概念、形成機理以及如何表征
使用Keithley 4200-SCS半導體表征系統進行氧化物可靠性測試
金屬化薄膜電容氧化時方阻會變大嗎
鎳氫電池的負極材料是什么

研究人員開發出高性能p型非晶氧化物半導體
金屬氧化物壓敏電阻的沖擊破壞機理&高能壓敏電阻分析
金屬氧化物壓敏電阻 (MOV) 概述:工作和應用
現代互補金屬氧化物半導體 (CMOS) 模擬多路復用TMUX734xF數據表

互補金屬氧化物半導體 (CMOS) 多路復用器TMUX4827數據表
具有閂鎖效應抑制特性的互補金屬氧化物半導體 (CMOS) 開關TMUX7236數據表
Littelfuse宣布推出SM10金屬氧化物壓敏電阻系列
簡單認識功率金屬-氧化物-半導體場效應管





 工商網監
工商網監
評論