 銥酸鹽開放骨架衍生的高活性長壽命析氧電催化劑
銥酸鹽開放骨架衍生的高活性長壽命析氧電催化劑
01
導讀
酸性析氧反應是幾種重要的電能-化學能轉換的基礎,這種能量密集型過程在工業上依賴于銥基電催化劑。以銥酸鹽為代表的的電催化劑性能強烈依賴于初始銥酸鹽的晶體結構和缺陷。最近報道的具有無定形活性相的銥酸鹽衍生的電催化劑通常表現出比結晶IrO2納米催化劑高10倍以上的活性,但結構穩定性更差(或銥浸出更嚴重)。并且,它們中的大多數在電催化條件下僅保持不到50小時的高催化活性。因此,希望開發新的銥酸鹽(預)電催化劑,其可以避免傳統催化劑中的從結晶到無定形的相變,從而實現催化活性和壽命的同時提高。
02
成果簡介
鑒于此,吉林大學鄒曉新教授團隊報道了具有開放框架結構的亞穩態銥酸鍶的相選擇性合成,該開放框架銥酸鹽衍生的納米催化劑在酸中給出了與最活躍的銥基析氧電催化劑相當的催化活性,并且保持其催化活性超過1000小時。文章以題為“Highly Active, Long-Lived Oxygen Evolution Electrocatalyst Derived from Open-Framework Iridates”發表在Advanced Materials上。
03 關鍵創新
(1)報道的開放框架銥酸鹽衍生的納米催化劑在酸中給出了與最活躍的銥基析氧電催化劑相當的催化活性,并且保持其催化活性超過1000小時;
(2)亞穩態銥酸鍶的轉化包括兩個主要步驟:酸中的Sr2+/H+離子交換和電催化條件下的原位結構重排;
(3)開放框架的銥酸鹽具有在酸中進行快速質子交換而沒有框架無定形化的能力。在酸性析氧過程中,所得質子化的銥酸鹽進一步重構為超小的、表面羥基化的、(200)晶面取向的金紅石納米催化劑,而不是普通的無定形IrOxHy相。這種微結構特征有利于電催化循環中羥基的氧化和O-O鍵的形成。
04
核心內容解讀
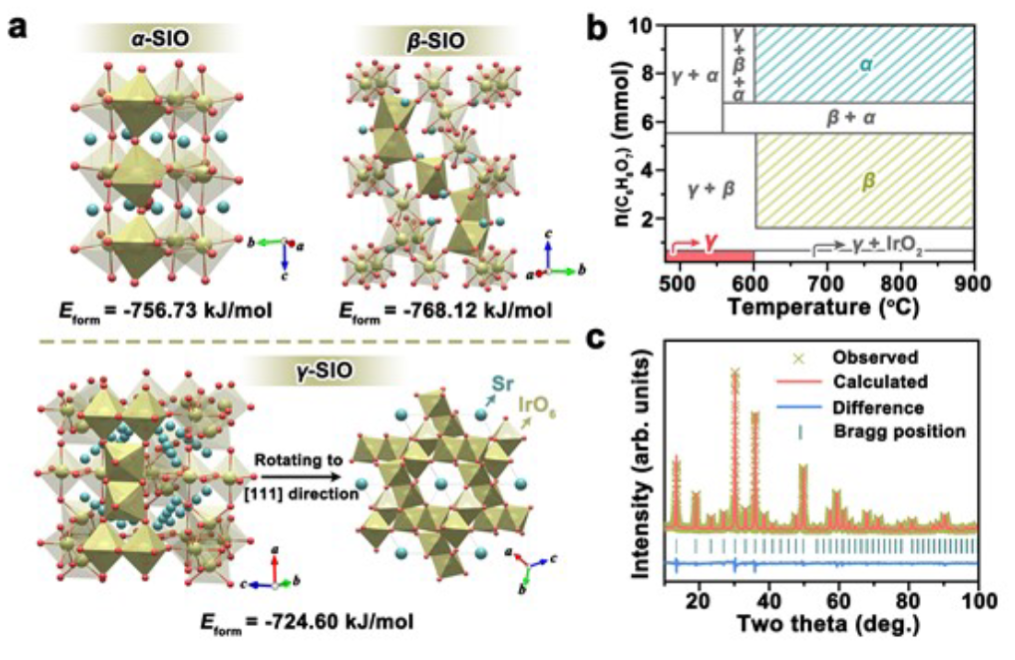
圖1. (a)α-SIO, β-SIO and γ-SIO的晶體結構。(b)各種銥酸鍶相的選擇性合成的示意圖。(c)具有γ-SIO細化圖的XRD圖。
圖1a顯示了三種銥酸鍶相的晶體結構。α-SIO和β-SIO都是致密結構,γ-SIO采用三維體心開放框架結構。α-SIO僅包含角共享IrO6八面體,而β-SIO由角共享IrO6八面體和面共享IrO6八面體二聚體組成。γ-SIO由共享邊緣的IrO6八面體二聚體組成。
如圖1b所示,與α-SIO和β-SIO的合成相比,用少量檸檬酸和低煅燒溫度實現了相純γ-SIO的合成。粉末x射線衍射(XRD)圖(圖1c)證明了具有開放框架結構的高純度銥酸鍶的成功合成。
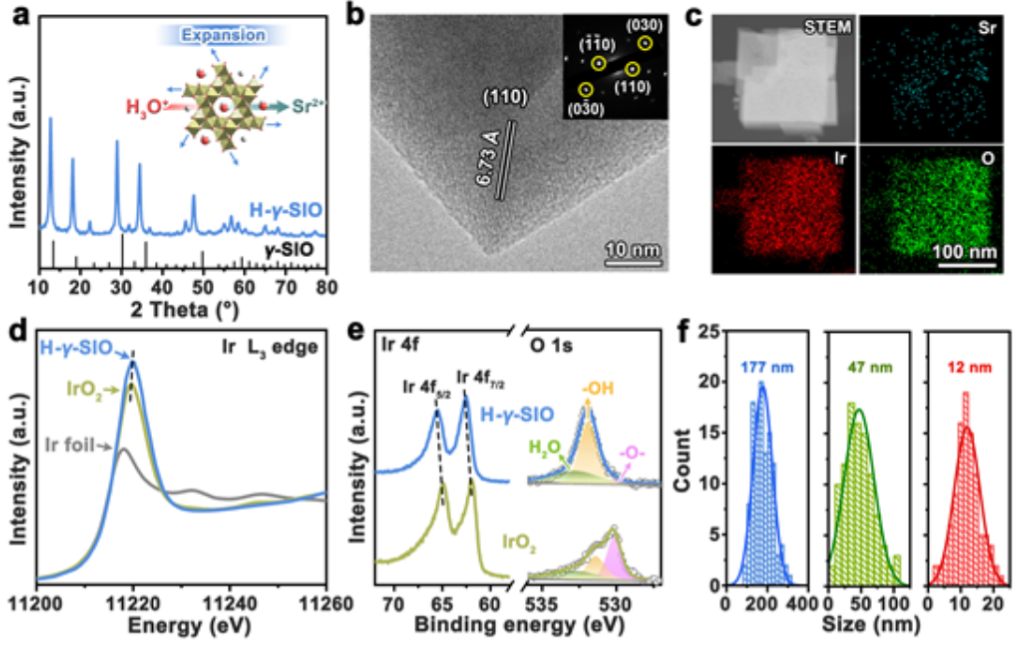
圖2. (a)H-γ- SIO的XRD圖。(b)H-γ- SIO的高分辨率透射電子顯微鏡圖像。(c)H-γ-SIO的相應元素映射圖像。(d)Ir箔、IrO2和H-γ-SIO的XANES光譜。(e)IrO2和H-γ-SIO的Ir 4f和O1s XPS光譜。(f)分別為H-γ-SIO-3、H-γ-SIO-2和H-γ-SIO-1的尺寸分布。
酸腐蝕對酸性OER電催化劑的結構有很大影響。在電催化研究之前,我們首先測試了γ-SIO在酸性溶液中的結構穩定性。在酸處理后,發現開放骨架中的Sr2+離子與H+離子完全交換,產生質子化形式(記為H-γ-SIO)。
H-γ-SIO的XRD圖(圖2a)與γ-SIO的粉末衍射圖非常吻合,表明H-γ-SIO保持了γ-SIO的開放骨架結構,但呈現出輕微的晶格膨脹。HRTEM圖像(圖2b)進一步證實了晶格膨脹。H-γ-SIO的元素映射圖像(圖2c)顯示了非常弱的Sr信號。經過酸處理后,γ-SIO中的Sr2+離子幾乎完全被H+離子交換。比較了H-γ-SIO、IrO2和Ir箔的Ir L3-edge X射線吸收近邊結構(XANES)光譜(圖2d)。
與IrO2相比,H-γSIO中Ir原子周圍的電子密度稍低。此外,相對于IrO2,H-γ-SIO的Ir 4fXPS光譜(圖2e)顯示出向更高結合能的小位移,并證明Ir在H-γ-SIO中的氧化態高于IrO2。與IrO2相比,H-γ-SIO具有高度羥基化的表面,這是Sr2+/H+離子交換的結果。
圖2f給出了三個樣品的尺寸分布和平均直徑(數均)分別為177、47和12 nm。相應地,這三種樣品按粒徑降序分別標記為H-γSIO-3、H-γSIO-2和H-γSIO-1。
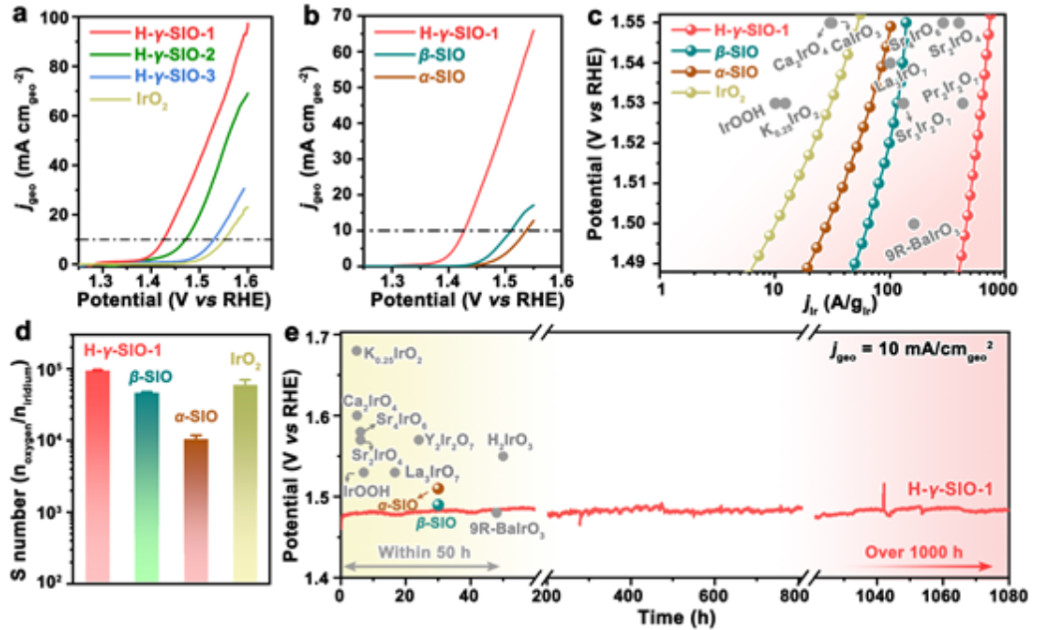
圖3. (a) H-γ-SIO-1、H-γ-SIO-2、H-γ-SIO-3和IrO2作為電催化劑的OER在0.1M HClO4中的極化曲線。(b)OER與H-γ-SIO-1、β-SIO和α-SIO在0.1M HClO4中的極化曲線。(c)H-γ-SIO-1的Ir質量活性與一些先前報道的在酸中由銥酸鹽衍生的電催化劑的比較。(d)H-γ-SIO-1、β-SIO、α-SIO和IrO2的計算S數。(e)在沒有紅外補償的情況下,在10 mA cmgeo-2電流密度和H-γ-SIO-1存在時,OER的計時電位曲線。
對三種不同粒徑的H-γ-SIO樣品的電催化性能進行評估,并以IrO2納米催化劑作為參比材料進行了研究。對比OER在酸中的極化曲線(圖3a)。在10 mA cmgeo-2電流密度下,H-γ-SIO-1、H-γ-SIO-2、H-γ-SIO-3和IrO2的過電位分別為200、248、270和320mV。
如圖3b所示, H-γ-SIO-1的電催化活性(通過幾何電流密度評估)優于所有先前報道的銥酸鹽衍生的電催化劑。進一步將測量的電流相對于銥的質量進行歸一化。H-γ-SIO-1在1.5 V下給出466 A·g-1的電流密度,比IrO2(10.3 A·g-1)大45倍。H-γ-SIO-1的銥質量活性也高于那些代表性的銥酸鹽基(預)電催化劑。以上結果表明,H-γ-SIO-1是一種高效的銥基氧化物電催化劑。
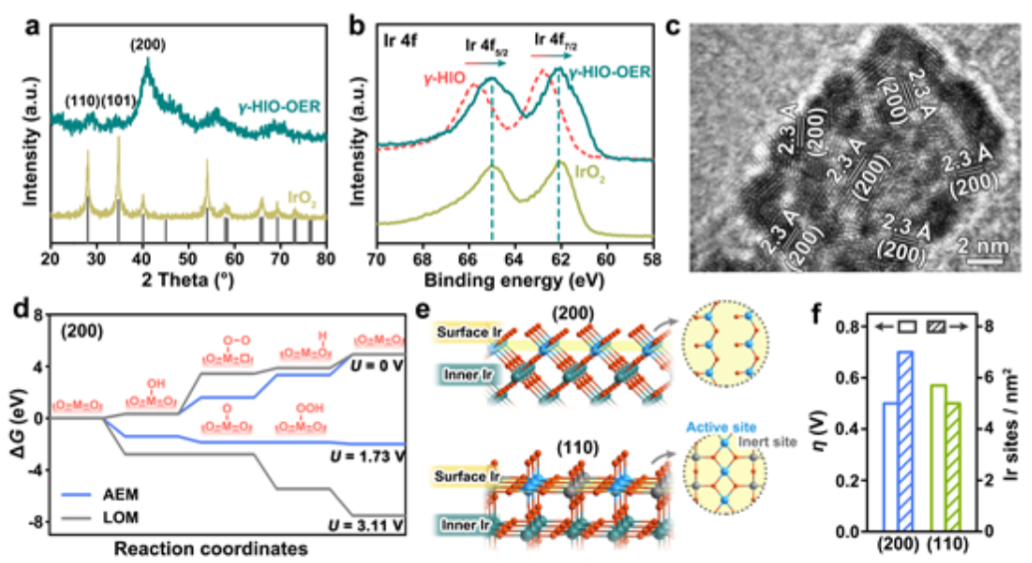
圖4. (a)在酸性溶液中以10 mA/cmgeo2進行10小時計時電位測試后,H-γ-SIO-1和IrO2的粉末XRD圖。(b)OER試驗前后,H-γ-SIO的Ir 4f XPS光譜。(c)試驗后H-γ-SIO-1的HRTEM圖像。(d)通過AEM反應途徑和LOM反應途徑,IrO2的(200)表面在不同外加電勢下的四級自由能圖。(f)IrO2的(200)和(110)表面的理論超電勢和I活性位數目的比較。
為了研究電催化過程中可能的結構重建,對OER后的H-γ-SIO-1進行了表征。與IrO2不同,H-γ-SIO-1的XRD圖在OER后發生了顯著變化催化后的H-γ-SIO-1(圖4a)的衍射圖與IrO2的衍射圖在峰位置上相同,并且最強的衍射峰是IrO2的(200)面,而不是常見的(110)和(101)面。H-γ-SIO-1的Ir 4f XPS光譜(圖4b)在OER后顯示出明顯的向較低結合能的移動,表明Ir的氧化態降低。催化后的H-γ-SIO-1的HRTEM圖像(圖4c)顯示樣品由晶格間距約為2.30的小納米顆粒(1~2 nm)組成,對應于IrO2的(200)晶面。H-γ-SIO-1在催化過程中經歷了重組,形成(200)取向的IrO2納米粒子。
基于OER后的上述表征,H-γSIO-1的優異催化穩定性可歸因于兩個方面。一方面,結晶金紅石IrO2納米顆粒是最終的活性相,其具有高的固有穩定性和在OER過程中超低的銥浸出。另一方面,制約析氧電催化劑在酸中長期穩定性的一個重要障礙是催化劑與集流體之間的界面接觸不良,導致催化劑/電極界面快速氧化鈍化,容易失活。另外,部分原因可能是其超小納米顆粒形態,改善了催化劑與集流體的界面接觸。
紅外基電催化劑上的OER包括兩種催化機理,吸附質析出機理(AEM)和晶格氧機制(LOM)。IrO2(200)表面上AEM和LOM的反應路徑的計算表明,類似于通常研究的IrO2納米催化劑,H-γ-SIO-1的OER優選遵循AEM途徑,而不是LOM途徑,因為其活性相是(200)晶面取向的IrO2。
根據AEM,我們通過密度泛函理論(DFT)計算研究了IrO2的(200)面上表面Ir位的催化活性。在四步反應機理中(圖4d), O–O鍵形成步驟(O*+ H2O →OOH* + H++ e-)是電勢決定步驟,并且超電勢被計算為0.5 V。金紅石IrO2的(200)和(110)表面上的IR活性位點的數量(圖4e)的計算結果表明,(100)表面(即(200)表面的等效面)比IrO2的熱力學最穩定的(110)表面具有更高的活性。
這些結果表明,IrO2的(200)表面具有更多數量的Ir活性位點,并且每個Ir活性位點的本征活性更高(圖4f)。這種不尋常的晶面取向和極小的納米晶尺寸使得H-γ-SIO-1在電化學測試中具有優異的OER活性。
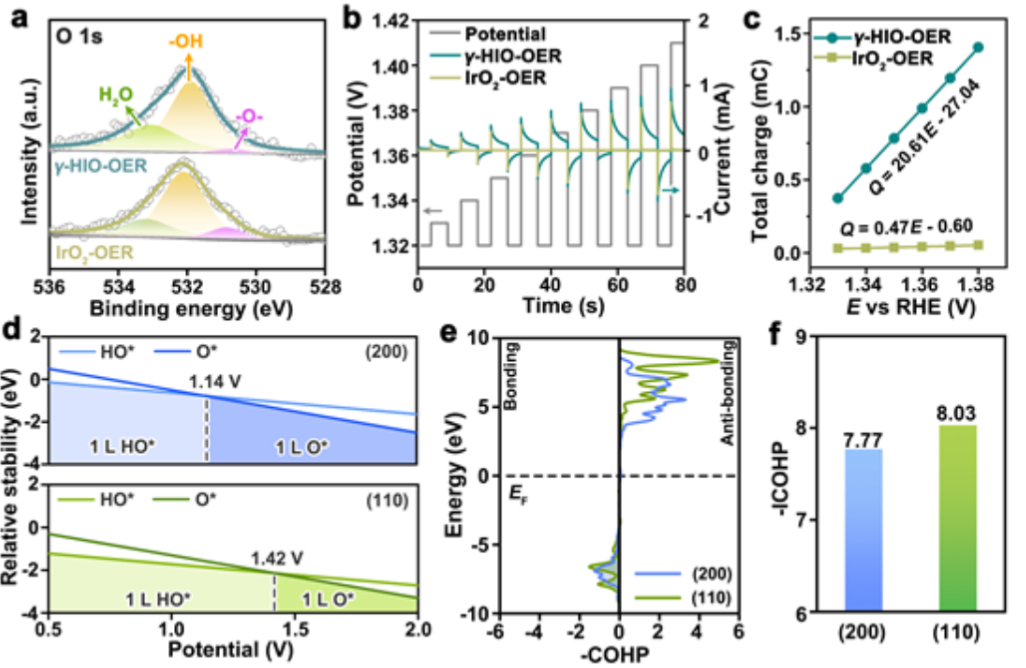
圖5. (a) 實驗后,H-γ-SIO-1和IrO2的O 1s XPS譜。(b)測量催化后的H-γ-SIO-1和IrO2對脈沖伏安法的電流響應。(c)儲存的電荷與催化后的H-γ-SIO-1和IrO2的電勢之間的關系。(d) IrO2的(200)和(110)表面的表面相圖。(e)O-H鍵的COHP曲線和(f)IrO2的(200)和(110)表面的積分COHP值。
基于上述催化機理,O*是決定OER活性的關鍵中間體。對于催化后的H-γ-SIO-1和IrO2樣品,它們的表面更傾向于OH-涂層。O 1s XPS分析(圖5a)反映催化劑表面儲存的氧化電荷的脈沖伏安法測量可用于測量羥基的去質子化能力。在不同的電壓脈沖下,催化的H-γ-SIO-1和IrO2顯示出交替的陰極和陽極電流脈沖(圖5b)。
通過對電壓脈沖的陽極電流響應進行積分,可確定儲存的氧化電荷(圖5c)。催化的H-γ-SIO-1的儲存電荷是IrO2的44倍。這些發現表明,與IrO2相比,催化的H-γ-SIO-1促進了O*中間體的產生,這是需要形成O-O鍵的限速步驟所必需的。
為了進一步理解去質子化能力,作者生成了IrO2的(200)和(110)表面的表面相圖(圖5d)。在低電位下,羥基封端的表面對IrO2的(200)和(110)表面都是最穩定的。然而,去質子化過程是由于在較高電位下,IrO2的(200)和(110)表面上的Ir位置優先被O*覆蓋。(200)面更容易產生O*,因為它的去質子化勢(1.14 V)低于(110)面(1.42伏)。
為了比較HO覆蓋的(200)和(110)表面的去質子勢,計算了O-H鍵的晶體軌道哈密頓布居(COHP)。結果(圖5e)顯示,O-H電相互作用由O2p和H1s態之間的軌道雜化主導。(200)表面的O-H雜化的積分COHP值(OH鍵強度的定量描述)為7.77,低于(110)表面的積分值(8.03)(圖5f),進一步表明,IrO2的(200)表面的O-H相互作用更弱,更容易去質子化。
05
成果啟示
開放骨架銥酸鹽在酸性電解質中電催化OER 期間重建為超小的、表面羥基化的、(200)晶面取向的金紅石納米催化劑。不尋常的(200)面上高活性Ir位點的大密度,加上超小的納米晶體尺寸,促進了對OER的優異催化活性。該發現為傳統的銥酸鹽電催化劑在酸性OER中面臨的從晶態到非晶態的相變問題提供了解決方案,并且可以鼓勵對高活性、長壽命的銥酸鹽基電催化劑的進一步研究。
審核編輯:劉清
-
DFT
+關注
關注
2文章
231瀏覽量
22752 -
LOMO
+關注
關注
0文章
2瀏覽量
7044 -
AEM
+關注
關注
0文章
25瀏覽量
10040 -
XRD
+關注
關注
0文章
133瀏覽量
9091
原文標題:AM:銥酸鹽開放骨架衍生的高活性長壽命析氧電催化劑
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
多功能高熵合金納米層實現長壽命無負極鈉金屬電池

燃料電池的主要材料 燃料電池的效率和性能

實時原位監測光電催化過程中反應物濃度與熱效應的微光纖傳感器技術

物聯網中高可靠低功耗長壽命繼電器方案_固態繼電器

網關助力催化劑產業升級,解決痛點問題!

浪潮通信信息榮獲2024 TM Forum催化劑項目大獎
芯伯樂CP2139直流馬達驅動:高效能、低噪音、長壽命的理想選擇
氧傳感器壞了有什么表現?氧傳感器常見故障有哪些?

相調控對鎳錫合金的電催化氮還原調控機制研究





 工商網監
工商網監
評論