 彈性界面相抑制氣體產生和促進鈉金屬負極均勻沉積
彈性界面相抑制氣體產生和促進鈉金屬負極均勻沉積
01
研究背景
鈉電池負極在醚類電解質中表現出優異的性能,優于酯類電解質。然而,其中的機制尚不清晰,而揭示該機制有助于診斷酯類電解質循環不良的原因,并優化電解質組成,促進鈉電池循環的可逆性。造成性能差異的一個重要因素是醚類電解質衍生固體電解質間相(SEI)層的彈性增強。然而,為什么SEI彈性增強會改善負極循環穩定性仍未得到深入研究?
02
成果簡介
近日,牛津大學Peter G. Bruce教授和華威大學Alex W. Robertson教授在Energy & Environmental Science上發表了題為“The role of an elastic interphase in suppressing gas evolution and promoting uniform electroplating in sodium metal anodes”的論文。
該工作通過使用原位電化學透射電子顯微鏡(TEM)對循環中的電極-電解質界面進行實時成像,揭示了彈性SEI如何阻止金屬負極界面上的氣體生成。高空間分辨率的TEM成像顯示,在酯類電解質鈉電剝離過程中,界面處迅速形成氣泡,而在醚類電解質中沒有觀察到這種現象,這些氣泡阻礙了鈉的完全溶出,并導致SEI從電極上脫落。
質譜測量結果表明,這種非共形的、不可彎曲的SEI必須不斷發生變化,導致形成SEI導致的鈉損失增加。醚類電解質中更有彈性的界面相能夠更好地保持與電極的共形接觸,防止氣體的形成,有利于形成平坦的電鍍形貌。
03
研究亮點
(1)本工作利用原位電化學TEM揭示了使用酯類和醚類電解質鈉時電鍍和剝離行為的差異。
(2)高分辨率成像顯示,酯類電解質形成了明顯的氣泡,在電剝離過程中,金屬-電解質界面處有強烈的氣體析出,導致SEI從金屬表面剝離。在醚類電解液中循環時,界面處未觀察到氣泡形成。
(3)原子力顯微鏡(AFM)表征表明,醚類電解質形成的SEI能夠在循環過程中更好地保持與電極的共形接觸,限制了氣體的產生,并最大限度地減少了SEI重組導致的鈉損失。
04
圖文導讀
1 M NaPF6溶解于碳酸乙烯(EC)和碳酸二甲酯(DMC)中(EC:DMC=1:1),作為酯類電解質, 1 M NaPF6溶解于二甲氧基乙(DME)中作為醚類電解質。為了更好地理解鈉在酯類電解質中的降解機制,還對添加了氟代碳酸乙烯(FEC)和碳酸乙烯(VC)添加劑(10%)的酯類電解質進行了研究。測定了Na||銅電池的電化學性能和阻抗行為。
圖1a顯示,酯類電解質電池產生了24%的低首圈庫侖效率(CE)。VC或FEC添加劑的加入顯著提高了首效,但仍然只能分別達到70%和74%。相比之下,醚類電解質電池產生了94%的CE,超過了所有的酯類電解質。在10次循環之后(圖1b),酯類電解質電池性能沒有明顯變化,無添加劑、VC添加劑和FEC添加劑電池的CE分別為22%、73%和75%。在10次循環后,醚類電解質CE穩定在99.8%以上(圖1b)。電化學阻抗譜(EIS)測量結果(圖1c)顯示,醚類電解質的離子電導率顯著提高。
為了診斷初始SEI形成對Na損失的貢獻,并量化SEI的大小和組成,對循環后的電池進行了在線質譜分析(在線MS)。圖1d-g顯示,在第一個循環之后,酯類電解質中大量的Na流失到SEI中(圖1d)。無添加劑、VC添加劑和FEC添加劑電解質中SEI造成的鈉損失分別為34%、21%和19%。采用醚類電解質能夠將SEI的損失量降低到3%,不到酯類電解質相對Na損失量的十分之一。
醚類電解質很大程度上防止了因“死”Na而造成的Na損失,將其限制在3%(圖1d),甚至顯著優于含添加劑的電解質。在一個循環(圖1e-g)后,對所測產物進行分析,可以定量無機SEI產物NaH和有機SEI產物(CH2OCO2Na)2和NaOCO2R。
從圖1d中的數據可以看出,在酯類電解質電池中SEI產物的相對質量較大。界面相NaH是由金屬鈉與氫氣反應形成的,其可以阻礙Na+在電極界面的遷移。有機SEI產物可進一步分解為無機的Na2CO3。因此,酯類電解質SEIs中發現的大量Na和Na有機物造成了它們較差的電化學性能。NaH將提高SEI的絕緣特性(圖1c),有機物將導致SEI的持續溶解和形成,從而導致低的CE(圖1b)和大量SEI形成。相比之下,醚類電解質SEI保持化學穩定。
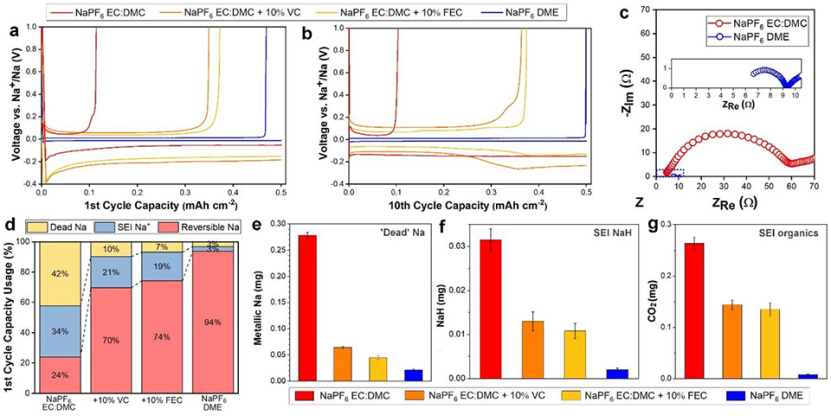
圖 1、在0.5 mA cm-2和0.5 mAh cm-2下,(a)第一次和(b)第10次充放電循環后,酯類或醚類電解質的電化學性能比較。(c)酯類和醚類電解質的EIS表征。(d)在線MS滴定法測量第一次充放電循環后Na的相對分布。一個循環后,在線MS定量測量(e)“死”Na, (f)NaH和(g)CO2的演變。
質譜測量表明,醚類電解質限制了“死”鈉的形成,表明它抑制了鈉枝晶。為了驗證這一點,使用原位 TEM直接捕捉醚類和酯類電解質的電沉積和溶解過程(圖2)。透射電鏡下的對比度取決于成像結構的相對密度。不幸的是,金屬鈉的密度非常接近周圍溶劑的密度,由于對比度低,特別是在醚類電解質的情況下,難以分辨沉積的金屬形貌。然而,仔細檢查醚類電解液的整個電鍍和剝離過程,還是能發現鈉的沉積和溶解基本均勻(圖2a)。
圖2b顯示了由鈉電鍍和剝離引起的強度變化。除了WE表面上均勻的鍍層外,還可以看到兩個微米大小的Na結構沉積和溶解,如圖2a中的箭頭所示。然而,在EC:DMC酯類電解質中,循環產生了更分散的沉積形貌(圖2c)。為了突出出現電鍍的區域,對第2幀(圖2d)圖像施加了背景減影,顯示出不規則的Na沉積形貌。
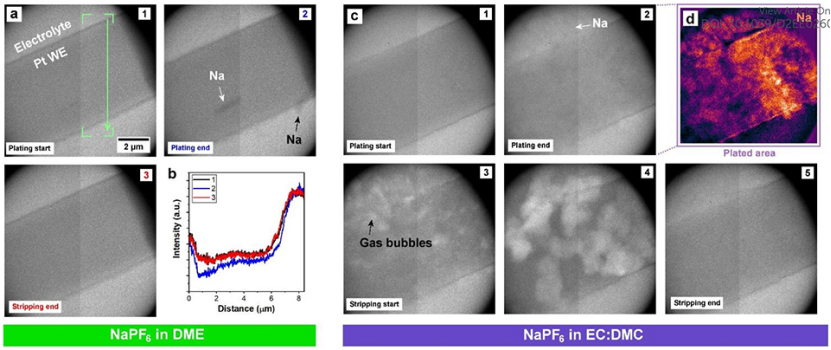
圖 2、原位電化學液體電池TEM成像。(a)NaPF6inDME電解液電池在10 mA cm-2下循環。(b)從圖1至圖3提取的強度分布圖。(c)NaPF6inEC:DMC電解液電池在10 mA cm-2下循環。(d)圖2減去背景后的彩色圖,突出Na沉積區域。
為了進一步探索酯類電解質體系中的局部氣泡形成,在添加10%FEC添加劑的EC:DMC電解質中,對循環后的電極進行了原位電化學高角度環形暗場(HAADF)掃描模式TEM(STEM)(圖3)。HAADF-STEM是一種暗場成像技術,與TEM成像相比,對比度倒置。例如,像氣泡這樣的低密度區域是黑暗的,而像Pt WE這樣的高密度區域是明亮的。在圖3b(白色箭頭)中可以初步分辨出微弱的Na沉積,隨著電鍍時間的延長,沉積量增加(圖3c)。
然而,一旦電剝離階段開始,將再次觀察到Na金屬表面的氣泡形成(圖3d,黃色箭頭)。有趣的是,這些氣泡向工作電極方向生長,膨脹并完全占據SEI殼層所包圍的空間(圖3g和h)。電剝離后,留下了明顯的SEI殼層,氣泡繼續生長,并被包含在其中。需要注意的是,由于SEI殼內的金屬鈉和電解質(圖3h中的深藍色)的對比度相似,不能區分它們,因此液體電解質可能在Na溶解后進入SEI殼內,隨后逐漸增大的氣泡將其取代。這些局部氣泡最終會消散(圖3e和f,綠色箭頭)。

圖 3、具有10% FEC 的1 M NaPF6inEC:DMC電解質中形成氣泡的原位電化學HAADF-STEM成像。(a-f)循環伏安掃描過程中不同電位下液體電池的STEM成像。(g)剝離過程中氣泡形成的放大圖。(h)為(g)的示意圖。(i)對應于圖a-f的循環伏安曲線。
為了進一步表征電鍍形貌的差異,并評估所形成SEI的力學性能,進行了AFM成像和納米壓痕測量(圖4)。與酯類電解質相比,醚類電解質電鍍的工作電極形貌更加光滑(圖4a和4c),表面粗糙度為26±5 nm,而非208±26 nm。剝離后(圖4b和4d),酯類電解質和醚類電解質的粗糙度分別下降到32±8 nm和6±2 nm。
原子力顯微鏡(AFM)圖像顯示,在酯類電解質中,金屬鈉的電鍍/剝離是非均勻的,在電極上形成粗糙的鈉島和凹坑。相比之下,使用醚類電解質第一次循環后,電極表面呈現出光滑的紋理,粗糙度甚至小于原始銅箔,證明了醚類電解質可以獲得良好的電鍍形貌。
原位AFM成像捕捉到了電鍍過程中粗糙度的演變,并揭示了醚類電解質形成平坦電鍍形貌背后的機制(圖4e-h)。電鍍600 s后,Na金屬均勻分布在Cu工作電極上。在進一步電鍍到1200 s時,觀察到金屬鈉在較低的位置生長,這些位置以黃色虛線區域突出,這些區域離對電極較遠。在電鍍結束(1800秒)時,同樣的現象也被觀察到,藍色虛線標記的區域在較低的位置再次出現金屬鈉的生長。Na的沉積傾向于優先發生在較低的位置,導致電鍍循環過程中,表面粗糙度減小(圖4g),這表明醚類電解質中的沉積機制保持了電極表面的平滑。剝離后電極的光潔度得以保持(圖4h),粗糙度為7.1±2.1 nm,證實了醚類電解質良好的剝離性能。
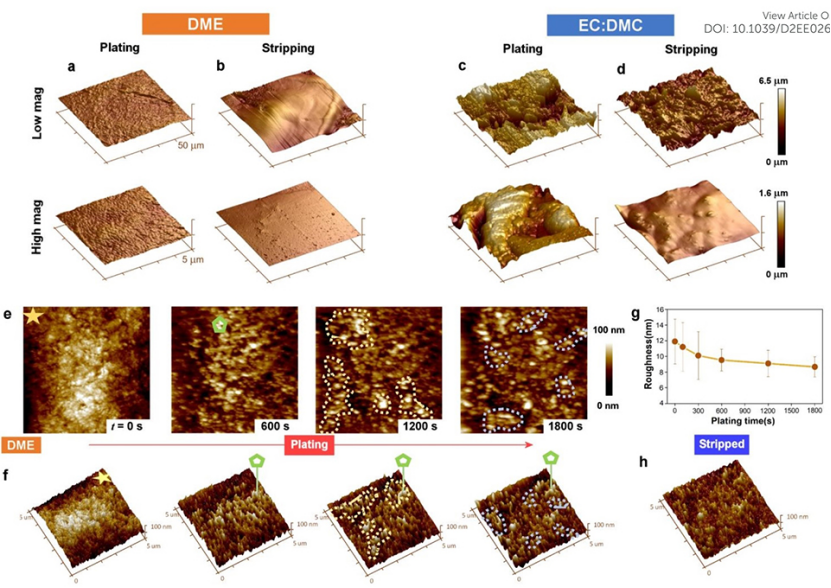
圖 4、(a-d)銅電極在醚類和酯類電解質中循環(0.5 mA cm-2@0.5 mAh cm-2)的AFM圖像,在(a,c)電鍍和(b,d)剝離后的成像。(e)0.5 mA cm?2下電鍍鈉過程中Cu電極的原位AFM形貌。(f)(a)中圖像的透視視圖。(g)電極粗糙度隨電鍍時間的演變。(h)恒流剝離至1V后的Cu電極。
AFM證實,醚類電解質衍生的SEI彈性性能增強,因此在鈉電剝離時沒有觀察到界面氣泡的形成。對于酯類電解質衍生SEI,剝離過程中Na金屬的溶解導致剛性SEI脫落,失去與負極的共形接觸,導致NaOCO2R和Na2CO3組分暴露在電解質中,隨后可能與NaPF6反應并釋放CO2氣體。而醚類電解質衍生的彈性SEI抑制了上述過程,因為它在剝離過程中與金屬鈉保持密切的共形接觸,而且它也由更少的NaOCO2R和Na2CO3組成。
為了證實這一機制,對與醚和酯類電解質中循環的鈉電池進行了差分電化學質譜(DEM)分析,并比較了產生的CO2量。在運行過程中,電池被密封,使產生的氣體積累起來,然后循環之后氣體被釋放并攜帶到質譜儀器中。結果表明,從酯類電解質電池中循環產生的CO2明顯多于從醚類電解質中循環產生的CO2(圖5)。
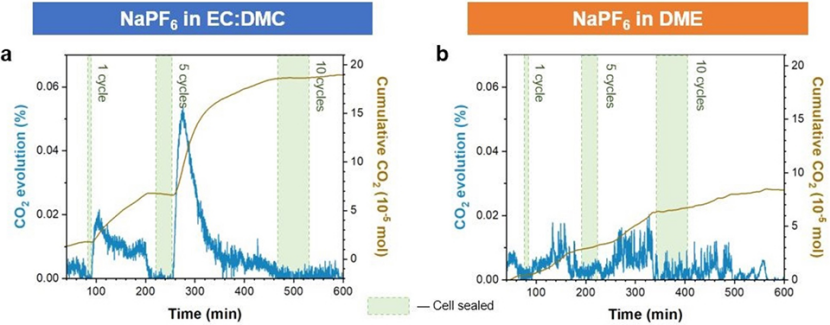
圖 5、差分電化學質譜(DEMS)比較酯類和醚類電解質電池的氣體析出。
05
總結與展望
本工作探索了醚類電解質中鈉負極性能提高的內在機制。原位電化學TEM顯示,在酯類電解液中,電極剝離時沿電極界面形成了大量氣泡。而在醚類電解液中循環時,沒有觀察到這種界面氣泡。這種在界面處形成的氣體會取代電解質,從而阻礙鈉的完全溶解。TEM成像和AFM也顯示,醚類電解質中電鍍的Na表面光滑,原位AFM表明,這是由于醚類電解質中形成的SEI更有彈性且更堅固。
這些良好的力學性能可以防止SEI在鈉剝離過程中發生反應,而非共形和脆性的SEI更容易從Na表面脫落,并發生副反應產生CO2。通過原位透射電鏡成像,SEI保持穩定,在電剝離過程中失去與電極的一致性,從而暴露出與電解質發生副反應的新區域。該工作表明,設計電解質以產生彈性和堅固的SEI層對于促進均勻平坦的鈉電鍍,并抑制產氣至關重要。
審核編輯:劉清
-
FEC
+關注
關注
0文章
40瀏覽量
13687 -
AFM
+關注
關注
0文章
59瀏覽量
20172 -
DME
+關注
關注
0文章
22瀏覽量
7411 -
固體電解質
+關注
關注
0文章
46瀏覽量
8389 -
鈉電池
+關注
關注
0文章
73瀏覽量
10234
原文標題:牛津大學EES:彈性界面相抑制氣體產生和促進鈉金屬負極均勻沉積
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦

高能鋰金屬電池中的宏觀均勻界面層與鋰離子傳導通道
硅碳負極生產的工藝流程

全固態鋰金屬電池的鋰陽極夾層設計
電氣設備放電產生什么氣體
降低半導體金屬線電阻的沉積和刻蝕技術
中微推出自研的12英寸原子層金屬鎢沉積設備Preforma Uniflex AW
詳解金屬偏析對焊點可靠性的影響

探索帶電鋰離子通道對金屬鋰負極枝晶生長的抑制作用


弱溶劑化少層碳界面實現硬碳負極的高首效和穩定循環

人工界面修飾助力高性能鋰金屬電池的最新研究進展與展望!





 工商網監
工商網監
評論