") 闡明Pt單原子催化劑的軸向配體效應(yīng)對(duì)堿性析氫反應(yīng)的影響
闡明Pt單原子催化劑的軸向配體效應(yīng)對(duì)堿性析氫反應(yīng)的影響
研究背景
電催化析氫反應(yīng) (HER) 制備氫氣是一種可有效解決碳排放問(wèn)題的方案。研究者對(duì)酸性介質(zhì)和堿性介質(zhì)中的HER展開(kāi)了廣泛的研究。在強(qiáng)酸性介質(zhì)中,為避免電極溶解,通常需要用貴金屬基催化劑電催化質(zhì)子還原。相比之下,多種催化劑在堿性電解質(zhì)中表現(xiàn)出較好的穩(wěn)定性。然而,堿性電解質(zhì)中的HER動(dòng)力學(xué)通常比酸性電解質(zhì)中低幾個(gè)數(shù)量級(jí)。此外,在堿性介質(zhì)中,催化劑表面結(jié)構(gòu)的敏感性也比在酸性介質(zhì)中高得多。因此,促進(jìn)堿性電解質(zhì)中水還原的緩慢動(dòng)力學(xué)對(duì)于降低綠色制氫的高過(guò)電位和相關(guān)能量損失至關(guān)重要。
開(kāi)發(fā)高活性單原子催化劑 (SACs) 是應(yīng)對(duì)上述挑戰(zhàn)的一個(gè)有效的解決方案。揭示化學(xué)/環(huán)境-催化性能的關(guān)系對(duì)于設(shè)計(jì)高活性的SACs至關(guān)重要。目前,調(diào)控SACs活性中心周圍化學(xué)環(huán)境的方法包括缺陷工程、退火、金屬載體相互作用、引入團(tuán)簇/納米粒子等。雖然利用上述合成策略開(kāi)發(fā)新材料取得了巨大進(jìn)展,但苛刻的條件不可避免地打破了SACs活性中心的同質(zhì)性,導(dǎo)致SACs活性中心的化學(xué)環(huán)境與催化性能之間的相關(guān)性的復(fù)雜性和不確定性。近年來(lái),一些Pt-SACs對(duì)堿性HER表現(xiàn)出良好的活性,但通過(guò)可控修飾單原子Pt位點(diǎn)周圍的化學(xué)環(huán)境來(lái)促進(jìn)HER活性的探索尚屬罕見(jiàn)。這促使研究人員設(shè)計(jì)Pt-SACs系統(tǒng)。在該系統(tǒng)中,Pt位點(diǎn)周圍的化學(xué)環(huán)境可以被精確操縱,從而確定可靠的構(gòu)效關(guān)系,并用于未來(lái)的催化劑設(shè)計(jì)。
成果介紹
新加坡國(guó)立大學(xué)Lei Wang和北京化工大學(xué)劉軍楓(共同通訊作者)通過(guò)電沉積的方法將Pt單位點(diǎn)引入NiFe層狀雙氫氧根 (LDH) 納米陣列上,發(fā)展了一種簡(jiǎn)單的輻照-浸漬方法來(lái)精確調(diào)整Pt-單位點(diǎn)上的軸向配體(如,?F,?Cl,?Br,?I,?OH),從而在保持Pt-SACs均勻性的基礎(chǔ)上建立了良好的化學(xué)-環(huán)境/HER-活性關(guān)系。通過(guò)詳細(xì)的光譜和電化學(xué)表征,表明Cl-Pt/LDH具有優(yōu)越的HER性能:在1.0 M KOH中,達(dá)到10 mA cm-2的過(guò)電位為25.2 mV。過(guò)電位為100 mV時(shí),質(zhì)量活性高達(dá)30.6 A mgPt-1,分別是HO-Pt/LDH和商用20% Pt/C的5倍和133倍。在相同的條件下,HER的活性遵循Cl-Pt/LDH》F-Pt/LDH》HO-Pt/LDH》Br-Pt/LDH》I-Pt/LDH的順序,證實(shí)了軸向配體對(duì)HER活性具有顯著的影響。密度泛函理論 (DFT) 計(jì)算表明,由于第一電子親和度高,Cl螯合Pt位點(diǎn)對(duì)OH*和H*都具有最佳的吸附親和力,從而促進(jìn)了緩慢的Volmer步驟 (水解離),這是堿性HER的典型動(dòng)力學(xué)限制步驟。此外,以Cl-Pt/LDH和NiFe-LDH分別作為陰極和陽(yáng)極催化劑,組裝了基于膜電極組件 (MEA) 的水電解槽。在60°C下,達(dá)到1 A cm?2需要的電壓為1.87 V,能量效率為80%。該研究證明了以原子精度設(shè)計(jì)SAC活性位點(diǎn)的重要性,為下一代催化劑的設(shè)計(jì)提供了理論支撐。相關(guān)工作以《Pinpointing the axial ligand effect on platinum single-atom-catalyst towards efficient alkaline hydrogen evolution reaction》為題發(fā)表在Nature Communications期刊。
圖文介紹
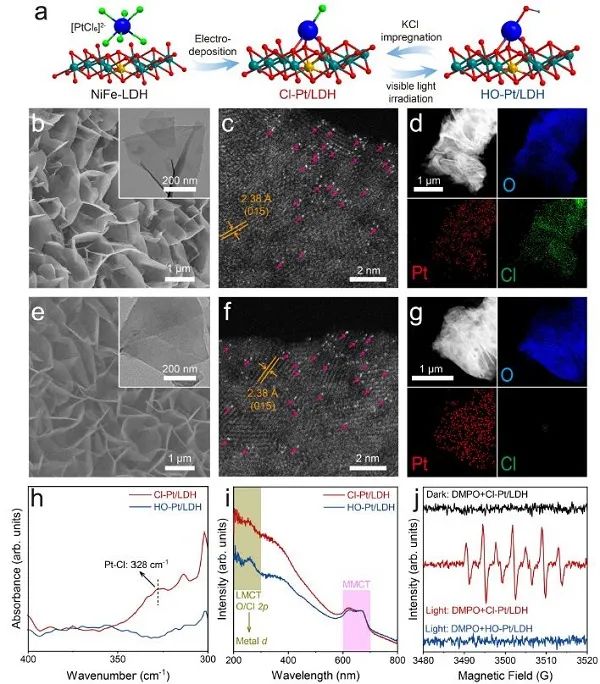
圖1. Cl-Pt/LDH和HO-Pt/LDH的合成過(guò)程和結(jié)構(gòu)表征。(a) 合成和配體交換過(guò)程的示意圖;(b,e) SEM (插圖:TEM);(c,f) HAADF-STEM,和(d, g) Cl-Pt/LDH和HO-Pt/LDH的元素映射;(h) Cl-Pt/LDH和HO-Pt/LDH的遠(yuǎn)紅外光譜和(i) UV-vis DRS光譜;(j)光/暗條件下Cl-Pt/LDH和HO-Pt/LDH的EPR光譜。
調(diào)整Pt/LDH軸向配體的輻照浸漬過(guò)程如圖1a所示。首先,通過(guò)水熱法合成了NiFe-LDH納米片陣列。然后,PtCl62-陰離子通過(guò)電沉積吸附到LDH表面,形成原子分散的Pt-位點(diǎn)。SEM和TEM圖像顯示,NiFe-LDH和Pt負(fù)載的LDH均呈現(xiàn)出橫向尺寸約為500 nm、厚度約為10 nm的片狀納米陣列 (圖1b,e)。通過(guò)像差校正的高角度環(huán)形暗場(chǎng)掃描透射電鏡(AC-HAADF-STEM,圖1c)未檢測(cè)到亞納米簇或納米顆粒,證實(shí)了Pt在Cl-Pt/LDH中以原子級(jí)分散。此外,Pt和O在納米板上的均勻分布表明Pt原子均勻地分散在LDH襯底上 (圖1d,g)。同時(shí),與NiFe-LDH的 (015 )面相對(duì)應(yīng)的晶格條紋的面間距約為2.38 ?,與XRD結(jié)果一致。為了交換Cl配體,用白光照射Cl-Pt/LDH (3.75 mW cm?2;30分鐘)。照射后Pt原子的原子分散沒(méi)有變化 (圖1f),但Cl信號(hào)的強(qiáng)度大部分減弱,表明Cl配體被成功去除。此外,輻照后遠(yuǎn)紅外光譜中329 cm?1處的信號(hào)減少進(jìn)一步證實(shí)了Pt-Cl鍵的丟失 (圖1h)。初步將Cl的損失歸結(jié)為OH- 作用下的陰離子交換,得到HO-Pt/LDH的產(chǎn)物,隨后用X射線吸附光譜法確定其結(jié)構(gòu)。在Cl-Pt/LDH和HO-Pt/LDH的紫外-可見(jiàn)漫反射光譜 (UV -vis DRS) 中,也觀察到配體-金屬電荷轉(zhuǎn)移 (LMCT) 誘導(dǎo)的吸收帶(~200 ~ 300 nm)發(fā)生了明顯的變化 (圖1i),而金屬-金屬電荷轉(zhuǎn)移 (MMCT) 沒(méi)有變化,說(shuō)明輻照后只發(fā)生了配體交換。如圖1j所示,當(dāng)Cl-Pt/LDH與自由基清除劑5,5-二甲基-1-吡羅啉N-氧化物(DMPO)自發(fā)暴露時(shí),會(huì)產(chǎn)生自由基信號(hào),這可能是由于Cl配體通過(guò)光生空穴氧化成Cl自由基,然后被DMPO捕獲形成DMPO+●自由基。相反,在黑暗中觀察到Cl-Pt/LDH樣品沒(méi)有自由基信號(hào),而在光照下觀察到HO-Pt/LDH樣品符合預(yù)期。
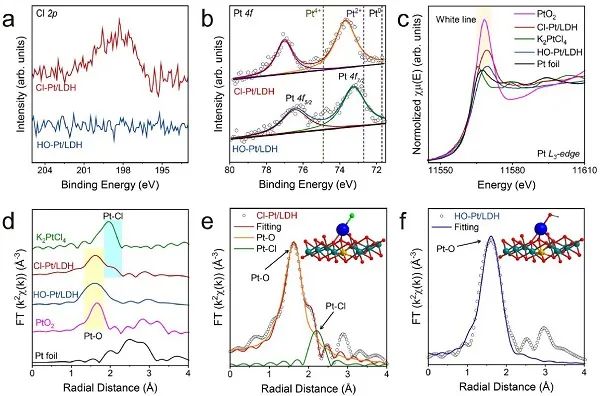
圖2.電子態(tài)和坐標(biāo)結(jié)構(gòu)的表征。(a) Cl 2p和 (b) Pt 4f的高分辨率XPS譜;Cl-Pt/LDH, HO-Pt/LDH, Pt箔,K2PtCl4和PtO2在Pt L3-邊的XANES光譜; (d) 對(duì)應(yīng)的FT-EXAFS光譜; (e) Cl-Pt/LDH和(f) HO-Pt/LDH的EXAFS擬合曲線。
采用XPS分析了Cl-Pt/LDH和HO-Pt/LDH的化學(xué)組成。光照后,Cl 2p信號(hào)消失為完全的配體交換提供了進(jìn)一步的證據(jù) (圖2a)。如圖2b所示,Cl-Pt/LDH和HO-Pt/LDH中Pt-原子的4f7/2峰都位于Pt2+ (72.7 eV) 和Pt4+ (74.9 eV)之間,說(shuō)明Pt-原子的平均價(jià)態(tài)在+2和+4之間。Cl-Pt/LDH中Pt 4f電子的結(jié)合能略高于HO-Pt/LDH中的結(jié)合能,這與先前觀察到電子從Pt轉(zhuǎn)移到Cl配體的結(jié)果吻合。用X射線吸收精細(xì)結(jié)構(gòu) (XAFS) 光譜研究了Cl-Pt/LDH和HO-Pt/LDH中單原子Pt位點(diǎn)的配位環(huán)境和電子結(jié)構(gòu)。如圖2c所示,Cl-Pt/LDH和HO-Pt/LDH的白線都位于K2PtCl4 (Pt2+) 和PtO2 (Pt4+) 的白線之間,說(shuō)明Cl-Pt/LDH和HO-Pt/LDH中Pt原子的價(jià)態(tài)在+2 ~ +4之間,與上述XPS結(jié)果一致。Pt在Cl-Pt/LDH和HO-Pt/LDH中的價(jià)態(tài)分別為2.78和2.08,說(shuō)明Pt-SACs的電子結(jié)構(gòu)存在強(qiáng)配體效應(yīng)。利用傅里葉變換(FT) k2加權(quán)擴(kuò)展X射線吸收精細(xì)結(jié)構(gòu) (EXAFS) 光譜進(jìn)一步分析了Pt-SACs的配位環(huán)境。如圖2d所示,在~2.50 ?處,Cl-Pt/LDH和HO-Pt/LDH均未檢測(cè)到Pt-Pt相互作用,說(shuō)明Pt原子在這些樣品中呈單原子分散狀態(tài)。此外,Cl-Pt/LDH在1.60 ?和2.06 ?附近有兩個(gè)明顯的峰,分別與PtO2的Pt-O路徑和K2PtCl4的Pt-Cl路徑有關(guān)。相比之下,HO-Pt/LDH中僅在~1.60 ?處有一個(gè)Pt-O樣峰,說(shuō)明光照后Cl配體被完全取代。
Cl-Pt/LDH的最佳擬合結(jié)果表明,F(xiàn)T-EXAFS譜中位于Pt l3-邊緣的1.60?處的主峰可以歸因于Pt-O第一配位,而在2.06 ?處的次峰可歸因于Pt-Cl第一配位 (圖2e)。根據(jù)Pt l3-邊的Cl-Pt/LDH的EXAFS擬合參數(shù),第一個(gè)配位球內(nèi)的O原子位于2.02 ?,配位數(shù)為3.02,表明Pt位為四面體。此外,在2.29 ?處的Cl原子估計(jì)配位數(shù)為0.93,被認(rèn)為是Pt上的垂直配體。綜合考慮所有因素,提出了Cl-Pt/LDH最可能的結(jié)構(gòu),如圖2e內(nèi)嵌所示。對(duì)HO-Pt/LDH的類似分析表明,它的第一配位球中只有O原子,總配位數(shù)為3.60,證實(shí)Cl配體被OH-取代 (圖2f),Pt位沒(méi)有發(fā)生其他變化。
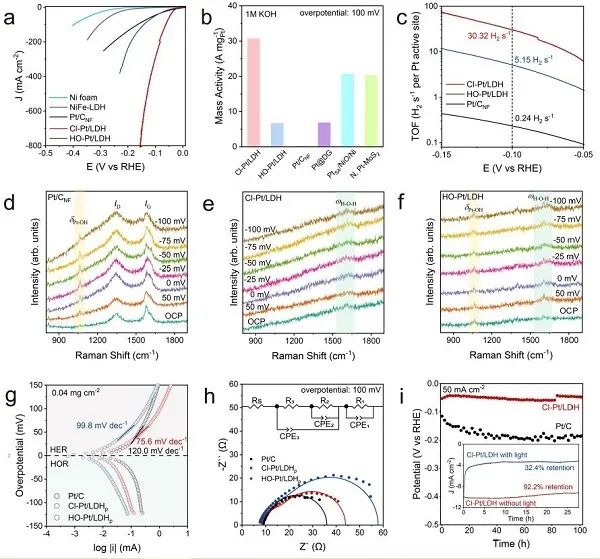
圖3. 堿性介質(zhì)中的HER性能。(a) 泡沫鎳、NiFe-LDH、Pt/C、Cl-Pt/LDH和HO-Pt/LDH的HER極化曲線;(b) 本研究中Pt基催化劑的活性,以及其他地方報(bào)道的最先進(jìn)的SACs: Pt@DG, PtSA/NiO/Ni和N, Pt-MoS2;(c) Pt基催化劑的TOFs圖;(d) Pt/CNF,(e) Cl-Pt/LDH和(f) HO-Pt/LDH的Operando拉曼光譜;(g) Pt/C, Cl-Pt/LDHp和HO-Pt/LDHp的微極化曲線;(h) Pt基催化劑的EIS Nyquist圖;(i) 陰極電流密度恒定為50mA cm?2時(shí),Cl-Pt/LDH和商用Pt/C的電位-時(shí)間曲線。
如圖3a所示,在所有測(cè)試的Pt-SACs中,Cl-Pt/LDH表現(xiàn)出優(yōu)越的HER活性和~100%的H2法拉第效率,達(dá)到10 mA cm?2、100 mA cm?2和200 mA cm?2的電流密度的過(guò)電位分別為25.2 mV、51.9 mV和72.3mV,優(yōu)于Pt/CNF (27.4 mV、164.9 mV和252.0 mV)和HO-Pt/LDH (41.5 mV、142.5mV和189.5 mV)。在100 mV過(guò)電位下,歸一化Pt負(fù)載時(shí)Cl-Pt/LDH的質(zhì)量活性估計(jì)為30.6 A mgPt?1(圖3b),顯著高于HO-Pt/LDH (6.6 A mgPt?1)、Pt/CNF (0.2 A mgPt?1)和其他論文報(bào)道的最先進(jìn)的Pt-SACs。此外,在過(guò)電位為100 mV時(shí),Cl-Pt/LDH (30.3 H2 s?1) 上Pt位點(diǎn)的翻轉(zhuǎn)頻率 (TOFs) 分別是HO-Pt/LDH (5.1 H2 s?1) 和商用Pt/CNF (0.2 H2 s?1)的5.9倍和126倍 (圖3c)。
采用原位拉曼光譜法研究了氧化還原過(guò)程中表面物種及其化學(xué)鍵的變化。如圖3d所示,在Pt/CNF上,HER僅在1062 cm?1處出現(xiàn)Pt-OH峰。因此,在催化條件下,*OH的解吸是緩慢的,這與之前的觀察結(jié)果一致。相反,在相同條件下,Cl-Pt/LDH上沒(méi)有觀察到這種明顯的Pt-OH峰 (圖3e)。在~1630 cm?1處只有細(xì)微的峰值,這可能是由于吸附水的H?O?H彎曲模式,表明Volmer步驟和隨后的*OH脫附步驟加快,導(dǎo)致HER動(dòng)力學(xué)增強(qiáng)。HO-Pt/LDH中也存在吸附水的細(xì)微峰值,這與HO-Pt/LDH較Pt/C活性增強(qiáng)相一致。注意,雖然HO-Pt/LDH上的HER中也發(fā)現(xiàn)了少量的Pt-OH峰(圖3f),但其強(qiáng)度不隨過(guò)電位的變化而變化,這表明?OH是軸向配體,而不是來(lái)自水還原。總之,Pt-SACs上HER活性的提高可能源于Volmer步驟的加速。在此條件下,Cl-Pt/LDHp的Tafel斜率 (76.5 mv dec?1) 明顯低于HO-Pt/LDHp (99.8 mV dec?1) 和Pt/C (120.0 mV dec?1),表明Cl-Pt/LDHp的遲緩Volmer步驟明顯增強(qiáng),RDS可能是Volmer步驟和Heyrovsky步驟的混合 (圖3g)。在相同條件下,在電流密度為50 mA cm?2 和500 mA cm?2時(shí),對(duì)Cl-Pt/LDH和Pt/C進(jìn)行了穩(wěn)定性測(cè)試 (圖3i)。在100小時(shí)的測(cè)試期間,Cl-Pt/LDH比商用Pt/C催化劑表現(xiàn)出了更好的耐久性,證明了Pt-SACs在堿性HER條件下化學(xué)結(jié)構(gòu)穩(wěn)定。
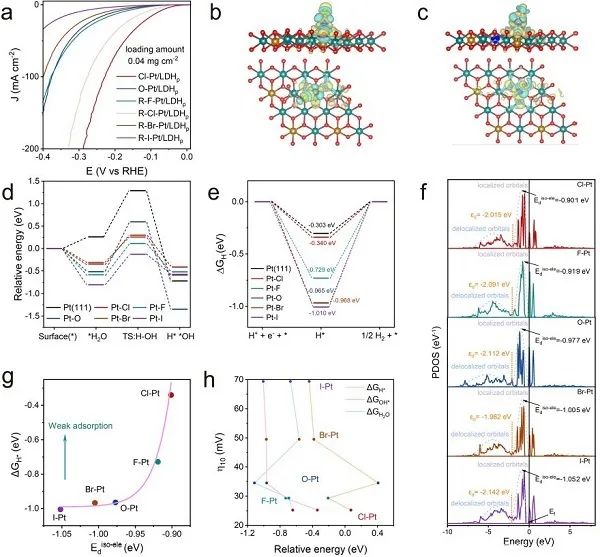
圖4. 軸向配體效應(yīng)及理論研究。(a) 不同軸向配體Pt-SACs的HER極化曲線;(b) Cl-Pt/LDH和(c) HO-Pt/LDH的計(jì)算模型和局域電場(chǎng)分布;(d) 計(jì)算水解離動(dòng)力學(xué)的能壘和 (e) H*在Pt-SACs表面的吸附自由能;(f) 計(jì)算Pt-SACs的Pt 5d能帶結(jié)構(gòu);(g) Eiso-ele d和ΔGH*的關(guān)系;(h) 堿性HER過(guò)電位 (在10mA cm?2) 與變量的關(guān)系。
不同軸向配體Pt-SACs的堿性HER活性隨鹵素原子的第一電子親和度而變化 (圖4a)。據(jù)EXAFS擬合結(jié)果建立了X-Pt/LDH (X = -F,-Cl,-Br,-I,-OH) 的計(jì)算模型。如圖4b,c所示,在Pt-X鍵合區(qū)域觀察到明顯的電荷重分布,這可能是HER活性增強(qiáng)的原因。對(duì)堿性HER在Pt-SACs上的逐級(jí)反應(yīng)能壘進(jìn)行了模擬,包括水解離的Volmer步驟、*OH的解吸以及隨后的H*轉(zhuǎn)化為H2。對(duì)于Volmer步驟 (圖4d),所有鹵素配位Pt/LDHs都比Pt(111)具有更強(qiáng)的水吸附能力和更大的水分解焓,導(dǎo)致Volmer步驟加速。其中Cl-Pt/LDH在Volmer步的能壘最小 (0.073 eV)。另一方面,Cl-Pt-位點(diǎn)在Pt-SACs中也表現(xiàn)出最優(yōu)的H結(jié)合能,導(dǎo)致H*轉(zhuǎn)化為H2的動(dòng)力學(xué)最佳 (圖4e)。注意,雖然Pt(111) 的H結(jié)合能比Cl-Pt/LDH略優(yōu)化,但它的水解離步驟可能較慢,導(dǎo)致整體HER動(dòng)力學(xué)較慢。綜合結(jié)果表明,H結(jié)合能并不是堿性HER的唯一描述符,OH結(jié)合能也起著重要作用。隨后,計(jì)算了孤立電子(Eiso-ele d)占據(jù)的窄Pt-5d軌道的平均能級(jí),分別為- 0.901,- 0.919,- 0.977,- 1.005和- 1.052 eV (圖4f)。這與上述XANES的結(jié)果一致,在Cl-Pt/LDH中,由于Cl配體的第一電子親和度最大,Pt的價(jià)態(tài)最高。根據(jù)d波段理論,表面中間體的吸附性能與催化劑的電子結(jié)構(gòu)直接相關(guān)。配體在Pt單原子上的第一電子親和度的提高可能會(huì)增加Eiso-ele d,從而進(jìn)一步減弱對(duì)氫的吸附。Eiso-ele d最高的Cl-Pt與H*中間體的相互作用最弱,因此H*更容易解吸附并形成H2 (圖4g)。總的來(lái)說(shuō),計(jì)算結(jié)果與實(shí)驗(yàn)觀察一致,用軸向配體修飾Pt單位點(diǎn)的電子結(jié)構(gòu)是調(diào)節(jié)HER活性的有效策略。綜上所述,X-Pt/SACs的堿性HER活性隨著配體第一電子親和度的增加而增加,因?yàn)?H和*OH的吸附能都是堿性介質(zhì)中HER活性的主要描述符 (圖4h)。
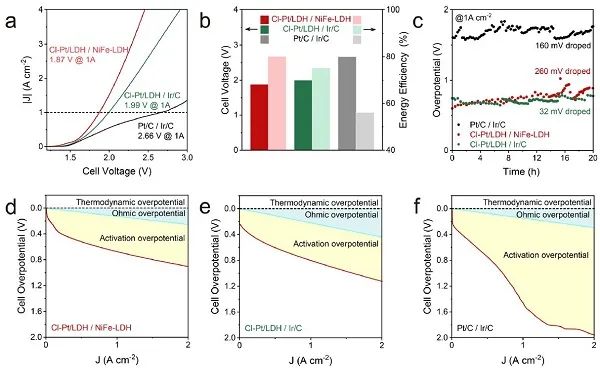
圖5. 在MEA電解槽中電催化HER。(a) 分別以Cl-Pt/LDH / NiFe-LDH、Cl-Pt/LDH / Ir/C和商用Pt/C/Ir/C為陰極和陽(yáng)極的MEA電解槽的LSV曲線;(b) 圖5a中電流密度為1 A cm?2時(shí)的電壓和能效;(c) 在60 °C下,電流密度為1 A cm?2時(shí),MEA電解槽電解水的穩(wěn)定性試驗(yàn);(d) Cl-Pt/LDH/NiFe-LDH;(e) Cl-Pt/LDH/Ir/C;(f) 商用Pt/C/ Ir/C的歐姆過(guò)電位和活化過(guò)電位;Cl-Pt/LDH和Pt/C的Pt負(fù)載分別為1.01 × 10?2和0.8 mgPt cm?2。
為了評(píng)估Cl-Pt/LDH作為陰極催化劑在實(shí)際操作條件下的適用性,以NiFe-LDH為陽(yáng)極,構(gòu)建了基于陰離子交換膜的膜電極組件 (MEA) 電解槽。建立了以Pt/C和Ir/C為基礎(chǔ)的MEA電解槽,并進(jìn)行了對(duì)比試驗(yàn)。在60°C下獲得了電流-電壓曲線 (圖5a)。在沒(méi)有進(jìn)一步優(yōu)化的情況下,基于Cl-Pt/LDH / NiFe-LDH的電解槽在電流密度為1.0 A cm?2 時(shí)的槽電壓 (1.87 V ) 比Cl-Pt/LDH / Ir/C (1.99 V) 和Pt/C / Ir/C (2.66 V) 的電解槽低得多。在此條件下,它們對(duì)應(yīng)的能量轉(zhuǎn)換效率分別為80%、75%和56% (圖5b)。如圖5c所示,Cl-Pt/LDH/Ir/C電解槽具有良好的穩(wěn)定性。從圖5d-f可以明顯看出,Cl-Pt/LDH/NiFe-LDH或Cl-Pt/LDH/Ir/C的活化過(guò)電位遠(yuǎn)低于Pt/C / Ir/C,這是由于Cl-Pt/LDH具有較好的HER活性,從而提高了能量效率。
總結(jié)
作者將Pt-SACs錨定在具有不同軸向配體 (?F,?Cl,?Br,?I,?OH) 的NiFe-LDH納米陣列上,通過(guò)簡(jiǎn)單的輻照浸漬過(guò)程制備了高活性堿性HER電催化劑。Cl-Pt/LDH在Pt- SACs和商用Pt/C中表現(xiàn)出最高的HER活性。Cl-Pt/LDH對(duì)堿性HER活性的增強(qiáng)是由于軸向配體的第一電子親和度最大,對(duì)Pt單位點(diǎn)產(chǎn)生了強(qiáng)烈的電子效應(yīng)。DFT計(jì)算表明,在單原子Pt位點(diǎn)上引入軸向配體可以調(diào)節(jié)Pt的d軌道孤對(duì)電子的平均能級(jí),進(jìn)而調(diào)節(jié)H*和*OH的吸附能。最后,在基于MEA的堿性水電解槽中評(píng)價(jià)了Cl-Pt/LDH的反應(yīng)速率,獲得了高達(dá)80%的能源效率,證明了其在制氫方面的應(yīng)用前景。該工作凸顯了調(diào)控催化中心周圍化學(xué)環(huán)境的優(yōu)勢(shì),其合成策略和軸向配體效應(yīng)都可以用于指導(dǎo)未來(lái)SACs的設(shè)計(jì),提高大規(guī)模綠色制氫的性能。
審核編輯 :李倩
-
電極
+關(guān)注
關(guān)注
5文章
813瀏覽量
27210 -
電解質(zhì)
+關(guān)注
關(guān)注
6文章
810瀏覽量
20049 -
催化劑
+關(guān)注
關(guān)注
0文章
92瀏覽量
10294
原文標(biāo)題:Nature子刊:闡明Pt單原子催化劑的軸向配體效應(yīng)對(duì)堿性析氫反應(yīng)的影響
文章出處:【微信號(hào):清新電源,微信公眾號(hào):清新電源】歡迎添加關(guān)注!文章轉(zhuǎn)載請(qǐng)注明出處。
發(fā)布評(píng)論請(qǐng)先 登錄
相關(guān)推薦
燃料電池氧電極催化劑的研究
燃料電池上甲醇水蒸氣重整制氫研究進(jìn)展
堿性醇類燃料電池新型催化劑的研究
中國(guó)科大制備出34種單原子催化劑的方法
高活性生物質(zhì)碳負(fù)載Fe/Pt單原子雙功能催化劑開(kāi)發(fā)
一種高效評(píng)估復(fù)雜合金納米團(tuán)簇電催化析氫反應(yīng)活性
低結(jié)晶和異質(zhì)結(jié)構(gòu)AuPt-Ru@CNT像高效多功能電催化劑
析氫反應(yīng)(HER)電催化劑在電解裝置的廣泛應(yīng)用
利用可持續(xù)方法獲得綠色能源的電催化分解水制氫
應(yīng)變效應(yīng)對(duì)催化劑活性的影響
如何提高HEAs催化劑的催化活性和優(yōu)選設(shè)計(jì)研究
EnSM:鋰硫電池單原子催化劑的基礎(chǔ)、應(yīng)用和機(jī)遇
雙原子催化劑綜述:適用于能源和環(huán)境催化的雙原子催化劑





 工商網(wǎng)監(jiān)
工商網(wǎng)監(jiān)
評(píng)論