 氮氣還原耦合氫氣氧化實現連續流電化學合成氨
氮氣還原耦合氫氣氧化實現連續流電化學合成氨
氨是合成化肥、藥物和精細化學品的重要原料。目前,工業合成氨采用Haber-Bosch工藝,即氮氣和氫氣通過鐵基催化劑,在高溫高壓(350-450℃,150-200 bar)的反應條件下合成氨。Haber-Bosch 工藝被認為是20世紀最具影響力的技術成就之一。
2021年,通過Haber-Bosch工藝,全球氨產量高達1.82億公噸。其中,約70%的氨用于了化肥生產,全球約50%的糧食生產依賴于氨衍生的化肥(Chem Catalysis, 2 (10), 2590-2613 (2022).)。以上足可見合成氨技術對人類的影響之大,然而目前工業合成氨路線,生產1公噸氨釋放約2.1公噸二氧化碳溫室氣體,而且需要大型工廠和高額的資本投資,該工藝的能耗就占全球能源消耗的1%,二氧化碳的排放量占全球排放總量的1.3%。
傳統的合成氨技術是密集型生產氨,化肥通過運輸到達用戶。目前,隨著可再生電力價格持續降低,以可再生能源為驅動力,直接從氮氣和水進行電化學合成氨是有潛力的合成氨路線之一。該過程可在較小規模的裝置中,利用分散的可再生能源進行分散式地合成氨,預期會帶來巨大的經濟和社會效益,例如降低缺乏交通網絡或基礎設施的發展中國家和偏遠地區的化肥價格。
近年來,電化學合成氨和電催化合成氨得到了科學界廣泛的關注和探究,期望電化學合成氨路線能夠取代或補充Haber-Bosch工藝。遺憾的是,2020年,Douglas R. MacFarlane課題組詳細調研了127篇水系電催化合成氨的文章(Nat Commun 11, 5546 (2020).),氨的產量太低以致于不能確定所產的氨是否來自氮氣還原,目前來看,水系電催化還原氮氣合成氨路徑的可行性仍然懸而未決。
2019年,Ib Chorkendorff教授課題組通過定量同位素和嚴格的氣體純化實驗表明(Nature 570 (7762), 504-508 (2019).),在室溫下電化學合成氨的可靠途徑之一是鋰介導氮氣還原反應(Li-NRR)。 鋰介導合成氨最早可追溯到1930年,Fichter等人使用LiBr和LiCl的醇溶液進行電化學合成氨(Helv. Chim. Acta 13, 1228–1236 (1930))。
1993年,Tsuneto等人進行了優化(Chem. Lett., 851-854 (1993)),使用四氫呋喃(THF)和乙醇(99/1體積比)作為溶劑,LiClO4作為電解質。1994,Tsuneto等人通過增加反應器壓力,再次提升了氨的法拉第效率。自2019年以來,Li-NRR經歷了快速發展的時期。
2022年,在20 bar條件下,在單室間歇性反應釜中,總電流密度已經實現1A/cm2(Joule 6, 2083-2101 (2022))。2022年,在15 bar條件下,在單室間歇性反應釜中,氨的法拉第效率已經接近100%(Nature 609, 722-727 (2022))。
盡管取得了較大進展,所合成的氨中的氮原子來自于氮氣還原,但是所合成的氨中的氫原子來自于哪里?在已報道的Li-NRR研究中,幾乎所有的報道都在陽極氧化有機溶劑作為質子源,合成氨不能以消耗有機溶劑為代價!基于此,Karthish Manthiram課題組(Nat. Catal. 3, 463-469 (2020))最早提出在陽極側引入氫氣氧化反應(HOR)來提供可持續的質子源,但是并未證明氨中的氫來自于HOR,而且電解池電壓高達20-30V(未給出陽極電位),導致偽能量效率(EE)只有2.8%,該體系只能夠穩定5-8 min。
Douglas R. MacFarlane課題組也曾試圖正在陽極引入HOR(Science 372, 1187-1191 (2021)),在高壓反應釜中通入0.5 bar H2和19.5 bar N2使用離子液體作為質子穿梭劑(proton shuttle),然而在單室間歇性反應釜,不僅存在H2傳質限制,而且H2可以與金屬鋰反應形成氫化鋰,這阻礙了鋰在室溫下活化氮氣的能力。
目前為止,大部分的Li-NRR研究在單室間歇反應釜中進行,存在以下挑戰:
(1)氣體反應物傳質限制:N2和H2必須溶于電解液才能參與反應。因此,通常增加壓力來實現較高的產氨效率。
(2)間歇性產氨,難以放大生產。
(3)難以利用HOR作為可持續的質子源,H2會消耗金屬鋰。
在常溫常壓(1 bar和25 oC)下進行Li-NRR的挑戰如下:
(1)低產氨法拉第效率和能量效率,在單室間歇反應釜中,FE和EE都低于10%。
(2)犧牲溶劑作為質子源。
(3)在裝配氣體擴散電極的電解池中,穩定性極差。
基于這些挑戰和難題,丹麥科技大學Jens K. N?rskov教授和Ib Chorkendorff教授等人設計了有效面積為25 cm2的連續流電解池,采用規整孔尺寸的不銹鋼網(SSC)作為氣體擴散電極來避免傳質限制,氮氣還原(NRR)耦合氫氣氧化(HOR),在常溫常壓條件下,實現了61%的法拉第效率,13%的能量效率。
開發了在有機溶劑中穩定催化HOR的PtAu合金催化劑,PtAu合金催化劑極大降低了陽極電位,避免了溶劑的氧化。通過原位同位素質譜實驗(D2氧化),證明了氨中的氫來自于HOR,EtOH作為質子穿梭劑。
圖文介紹
首先構建了一個三室連續流反應器,GDE位于氣體室與電解液室之間,氣體反應物直接供給在GDE電極的一側,氣體流道的面積是25 cm2。Li-NRR在連續流電解池中的過程如下:鋰離子(Li+)通過固體電解質界面(SEI)從本體電解液擴散到陰極電極表面電化學還原為金屬鋰,隨后金屬鋰與N2反應形成氮化鋰 (LixNyHz),氮化鋰通過質子穿梭劑(EtOH)質子化來連續產生氨。PtAu陽極催化劑上穩定地發生HOR為該過程提供可持續的質子源。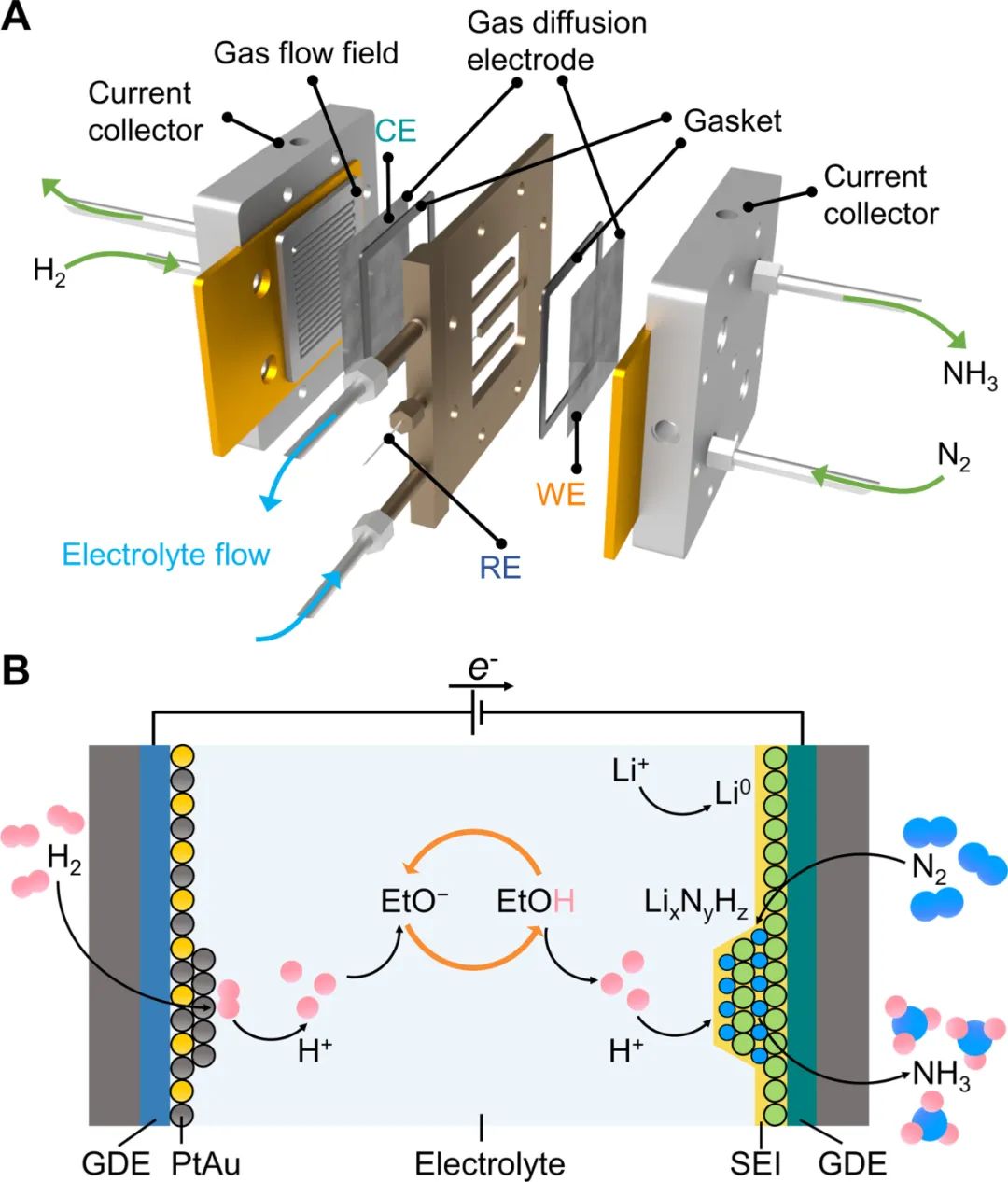
圖1. 用于電化學合成氨的連續流反應器的示意圖和Li-NRR過程示意圖
目前為止,極少數文獻研究了有機電解液中的HOR,與水系中的經典HOR不同,有機體系中的HOR催化劑非常容易失活,有機溶劑或反應中間體會毒化活性位點。在有機體系HOR中,Pt的催化活性在幾分鐘內失活。Douglas R. MacFarlane課題組探究了一系列在有機體系中的HOR催化劑,發現失活速率最小的PtRu催化劑在20 min左右也會失活(ACS Catal. 12, 5231-5246 (2022))。
因此,非常需要開發一種在有機體系中長期穩定的HOR催化劑。在小分子電氧化(甲酸/甲醇/乙醇)領域,Pt很容易被一氧化碳(CO)或其他中間體中毒。在Pt基雙金屬催化劑中,PtAu表現出良好的催化活性和抗CO中毒的特性。還有研究表明,用Au團簇修飾的Pt可極大增加氧還原反應的穩定性。受這些研究的啟發,作者認為Au對抑制有機分子或中間體的吸附有積極作用,PtAu催化劑有可能成為有機體系中最穩定的HOR催化劑之一。
理論計算表明,與純Pt表面相比,H、CO、THF和呋喃等物種在PtAu表面的吸附被削弱。H在PtAu表面上的吸附能比略弱于H在Pt表面的吸附能,從而使得PtAu的HOR反應速率比純Pt高1-2個數量級(圖2A)。與此同時PtAu表面上HOR相對于THF氧化反應的選擇性比純Pt表面高5-6個數量級,這顯著地降低了THF氧化副反應的速率。
更重要的是,CO作為毒化Pt基金屬的主要中間體,在Li-NRR反應中,其主要來源于乙醇氧化過程中碳碳鍵(C-C)的斷裂。理論計算表明,C-C鍵在純Pt表面斷裂所需要的反應能壘在室溫條件下極易克服,但是在PtAu表面,C-C鍵斷裂需要克服較高的反應勢壘所以不易發生, 從而抑制了CO的產生和表面活性位點的毒化。
通過氫氣泡模板法在SSC基底上制備了Pt和PtAu電極(Pt/SSC和PtAu/SSC)。HOR性能是使用1M LiBF4的THF電解液在氬氣手套箱中進行評估。在恒電流測試(CP)期間,Pt/SSC在幾分鐘內失活,陽極電位增加到1V vs. Pt,此電位下可發生THF氧化的副反應(圖2B)。
相比之下,PtAu/SSC在5小時內電位保持在0.3V vs. Pt,證明了PtAu在有機體系HOR中的高活性和長期穩定性(圖2B)。在對PtAu進行長期穩定性測試后,角分辨XPS結果表明Pt在PtAu催化劑表面富集(圖2C)。DFT計算表明,即使是微量的呋喃、CO和甲基也會導致Pt原子偏析到表面(圖2D),而且底層的Au進一步削弱了Pt覆蓋層對有機物的吸附強度。
底層的Au促進了表面類Pt(111)覆蓋層的形成,記作Pt(111)/Au。乙醇氧化中,C-C鍵斷裂所需要的反應勢壘在Pt(111)/Au表面的是在PtAu合金的1.5倍,而Pt(111)/Au上的HOR本征活性與PtAu相當。所以,PtAu作為真實催化劑Pt的前體,真實催化劑Pt(111)/Au表面在保有與PtAu合金相同的HOR本征活性同時,進一步抑制了CO的產生和表面活性位點的毒化。
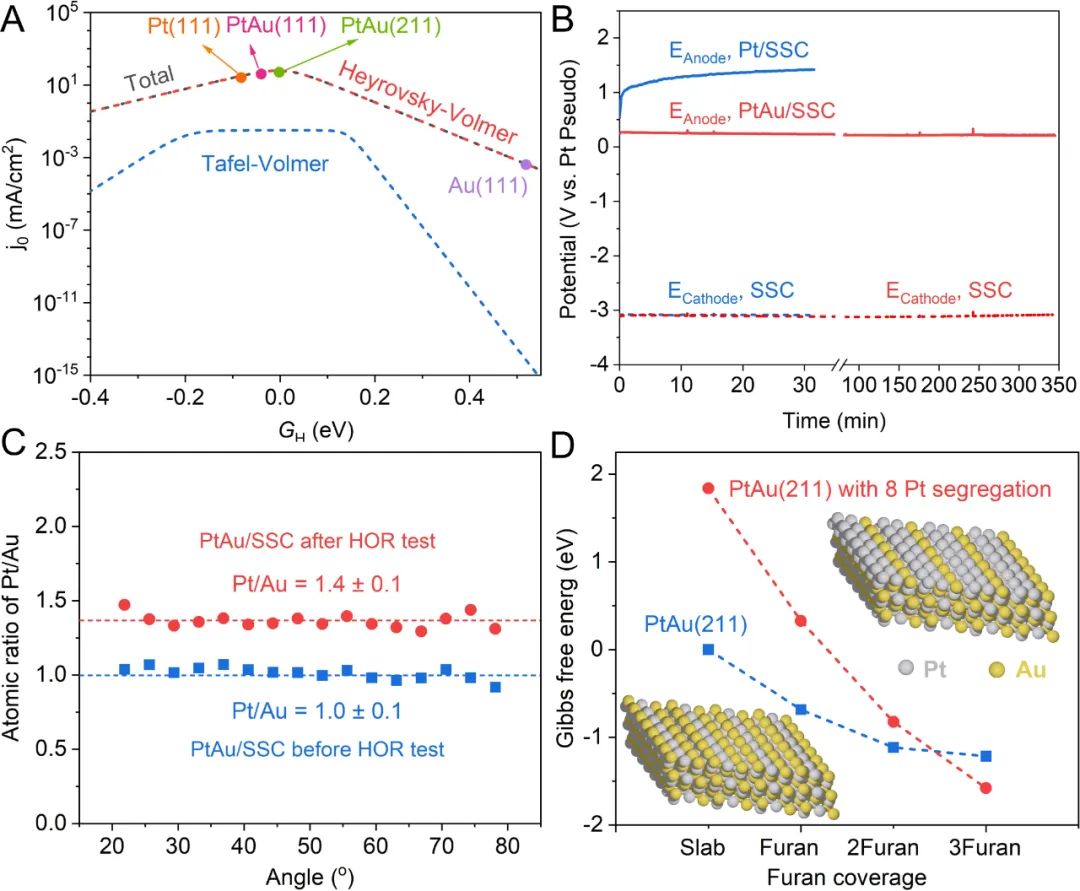
圖2. HOR催化劑的理論和實驗探究
為了證明NRR與HOR的耦合,在連續流電解池中進行了同位素原位質譜實驗。當D2流過陽極氣體室,測量陰極氣體出口處的產物,原位質譜可將電解液反應產生的含H的產物和D2氧化產生的含D的產物區分開來,最初,陰極完全被新鮮的電解液覆蓋,產生主要是含H的產物,如NH3和NH2D。
隨著實驗的進行,越來越多的含D產物形成(圖3C),最終完全氘代的氨(ND3)被檢測到。這有力地證明了NRR和HOR的耦合作用,所合成的氨中的氫來自于HOR,并證明了乙醇作為質子穿梭劑的能力。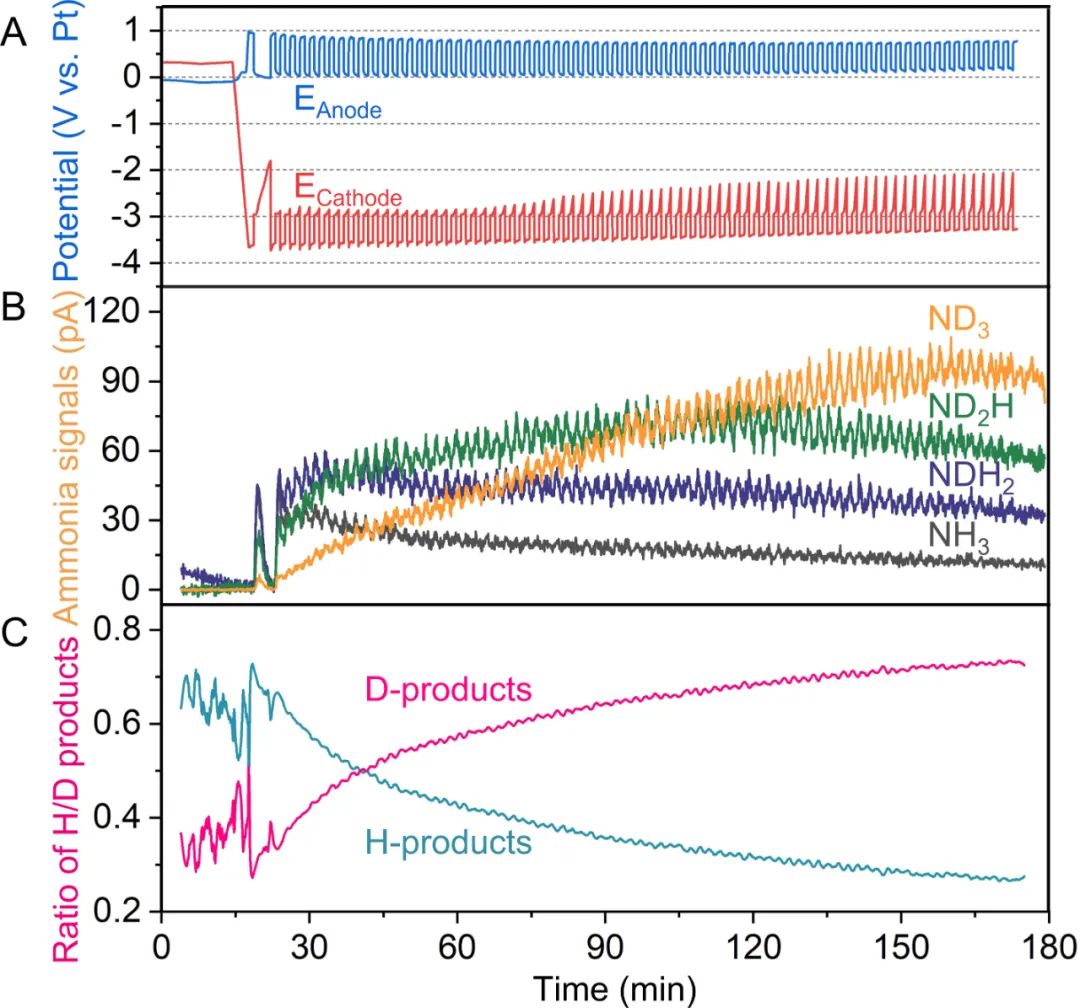
圖3. 同位素標記(D2氧化)原位質譜分析
采用電位循環(potential cycling)策略提高Li-NRR的性能,即在施加電位和開路電壓(OCV)之間切換電位。改變電位循環時間的實驗表明,最佳電位循環的條件是 1 min進行鋰沉積,1 min進行開路電壓(1 min deposition + 1 min resting)。如圖4 A所示,陰極的靜息電位大約是-2 V vs. Pt。
采用模數電池技術(modulo battery technique)可以控制靜息電位,改變靜息電位的實驗表明,最佳的靜息電位是-2 V vs. Pt。Li-NRR與陽極的HOR耦合,使得平均陽極電位保持在0.6 V 左右,極大降低了電解池的槽電壓(4.3 V)。在連續流電解池中評估質子穿梭劑(EtOH)濃度對性能的影響(圖4 B),最佳EtOH濃度為0.25 vol.%。
在最優條件下,使用30 μm SSC實現了61 ± 1%的產氨FE。電位循環的促進作用在質子和氮氣擴散限制區表現出相反的作用。這符合理論模型分析,在質子限制區,在電位循環的靜息期,沉積的鋰金屬與N2反應生成氮化鋰,氮化鋰溶解之后釋放NH3,從而提高性能。
在N2限制區,沉積的鋰金屬與質子反應形成氫化鋰,氫化鋰溶解產生H2,導致產氨的選擇性降低。所產的氨主要分布在氣相、電解液和電極沉積物中,最佳條件下,大約50%的氨分布在氣相中,這有利于后期分離和富集氨(圖4 C)。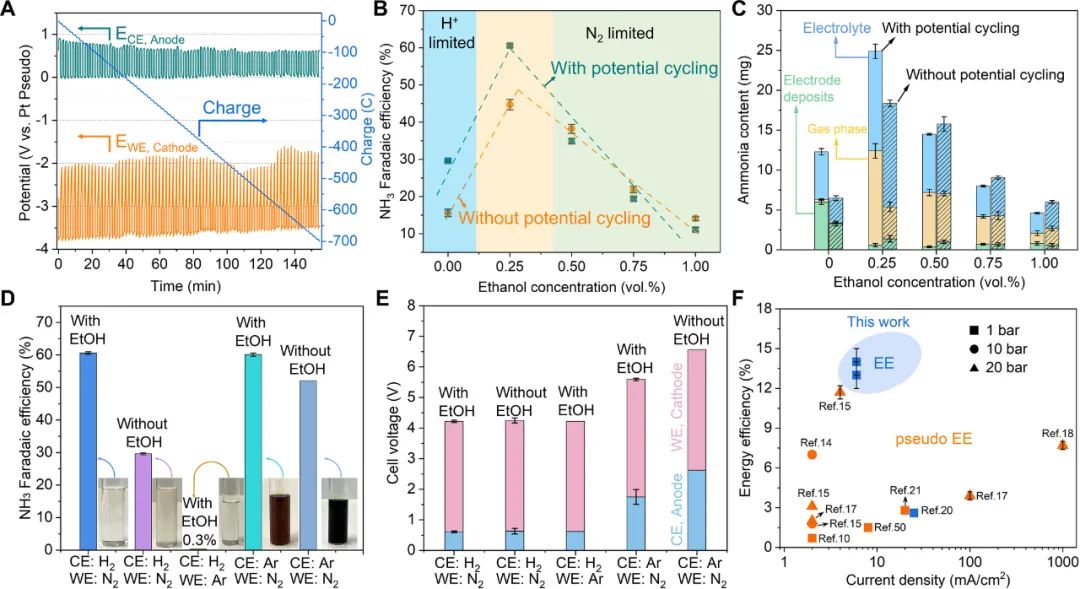
圖4. 電位循環法和質子/氮氣限制區域的權衡策略提高鋰介導合成氨的性能
對照實驗進一步確認HOR對陽極電位降低的重要性(圖4 D和E)。在典型的實驗條件下(陽極通入H2,陰極通入N2,使用EtOH作為質子穿梭劑),測試后的電解液無色透明,陽極電位為0.6 V。其他條件相同時,當在陽極測通入Ar時,測試之后的深色電解液表明有機溶劑嚴重的氧化和分解,陽極電位高達1.7 V (有EtOH)和2.6 V(無EtOH),這清晰地表明HOR可保護電解液不被氧化,降低了陽極電位。
采用NMR分析反應前后電解液的變化也支持了這一結論。圖4 F總結了電流密度和能量效率(EE),需要指出的是,偽能量效率未使用氫氣或者所計算的EE未包含產氫所需的能量。這項工作著重證明了在陽極引入HOR可大大降低陽極電位,證明了所產氨中的氫來自于HOR,不是犧牲有機溶劑。
還強調了整個系統能量效率以及在陽極上引入HOR提供可持續氫源的重要性。未來的工作應側重于在更高的電流密度下獲得高EE和FE,以提高產的氨速率。 只有當陽極真正使用氫氣或水作為可持續的氫源時,實現高FE和電流密度才具有實際的意義。
盡管該研究開發了有機體系中HOR高穩定性的PtAu催化劑,提高了流動電解池的運行穩定性,實現了連續化電化學合成氨,但是這項工作并未解決所有在工業應用層面的鋰介導合成氨的問題。未來的Li-NRR研究應該著眼于提高電流,優化H2/N2的傳質以及精確調節GDE上氣體和液體之間的壓力梯度。
主要目標應該是在中試規模的連續流電解池中,在工業電流密度條件下,實現高的FE和EE。可持續的氫氣可以通過可再生電力驅動的電解水獲得。循環陽極出口H2可能是提高陽極H2利用效率的途徑之一。本研究的這些發現為鋰介導合成氨的進一步發展提供了堅實的基礎和指導。
審核編輯:劉清
-
電解電容
+關注
關注
14文章
671瀏覽量
50823 -
ssc
+關注
關注
0文章
24瀏覽量
11205 -
電解池
+關注
關注
0文章
24瀏覽量
9517
原文標題:Science:氮氣還原耦合氫氣氧化實現連續流電化學合成氨
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
電化學氣體傳感器信號放大調試經驗
掃描速率對各體系的電化學行為有什么影響
哈爾濱工業大學/南方科技大學:聚焦離子束制備高分辨率電化學-電致發光耦合雙極納米電極陣列傳感器

電化學測試方法詳解
電化學儲能和電池儲能的關系
電化學儲能與物理儲能的對比
電化學儲能系統的組成與作用
電化學儲能的基本原理介紹
深度解析電化學儲能最新官方數據






 工商網監
工商網監
評論