

【研究背景】
在各種鋰離子電池負極材料中,硅在室溫下擁有3590 mAh g-1的超高比容量(約為石墨負極的10倍),被認為是最有前景的的負極材料之一。硫化物固態電解質在室溫下擁有杰出的離子傳導性并可以和電極材料機密接觸,從而為全固態電池帶了有優秀的性能。然而最近的報道發現硫化物固態電解質在硅負極電化學反應中并不穩定,而且碳的添加可能加劇這一過程。由于高達300%的體積變化,純硅負極在脫鋰后往往會出現微米級的空隙,這會帶來電極分層的風險。納米硅 (《100 nm) 表現出更高的結構穩定性,因為更小的尺寸可以潛在地減少應力的產生。但是納米硅具有低電子電導率和低離子擴散率,所以大多數報道的納米硅基負極是具有固態電解質和碳添加劑的復合負極材料。因此我們需要系統的了解添加固態電解質和碳添加劑對納米硅負極材料的電池性能、硫化物電解質的電化學穩定性、和電極結構的演變有何影響。
【工作介紹】
近日,美國東北大學Hongli Zhu教授、Juner Zhu教授,以及康涅狄格大學Hongyi Xu教授等人使用原位X射線吸收近邊結構(XANES)光譜、掃描電子顯微鏡(SEM)、以及X射線納米層析(XnT)技術系統地分析了不同納米硅負極(純納米硅、納米硅+固態電解質、納米硅+固態電解質+碳添加劑)的電化學和結構演變。原位 XANES 顯示硫化物電解質在硅負極中發生了電化學分解,而且碳的添加加速了這一過程;但是這種分解只在第一次鋰化過程中發生了,在隨后的循環中分解產物是穩定的,且沒有觀察到其對電池性能的明顯影響。SEM 和 XnT的結果顯示在納米硅負極中添加固態電解質和碳提高了其結構穩定性并減少了孔洞和空隙的產生。通過化學-彈性-塑性模型(chemo-elasto-plastic model)模擬表明,添加較軟的固態電解質和碳可以減輕硅體積膨脹引起的應力,從而提其高機械穩定性。權衡固態電解質和碳添加劑在增強反應動力學和結構穩定性方面的利處和其有限的在電化學穩定性方面的弊除,使用包含固態電解質和碳的納米硅復合負極表現出了更高的硅利用率,比純納米硅和僅具有固態電解質的納米硅復合負極具有更高的比容量和更好的倍率性能。該文章發表在國際頂級期刊Advanced Energy
Materials上。Daxian Cao, Tongtai Ji, Avtar Singh為本文第一作者。
【內容表述】
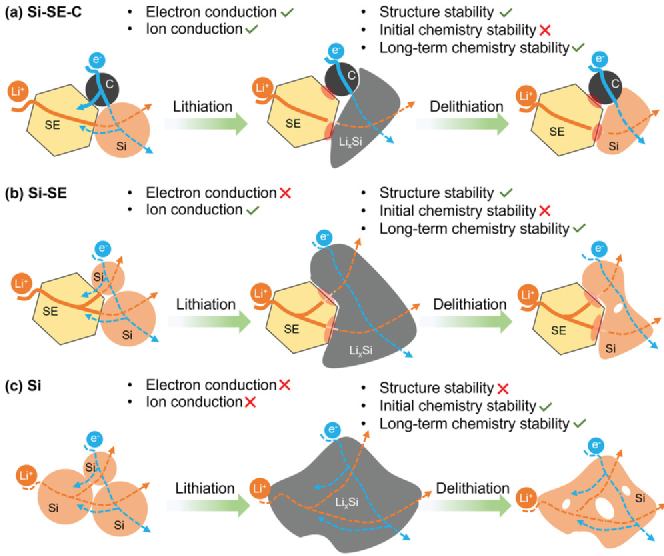
圖1示意圖說明了(a)納米硅+固態電解質+碳添加劑 (Si-SE-C), (b) 納米硅+固態電解質 (Si-SE)、和 (c) 純納米硅(Si)負極在全固態電池中的化學和結構演變中優缺點的總結和對比。在Si-SE-C中,三種成分在電池測試前是化學穩定的。C和SE分別在復合負極中建立了電子和離子傳導通路,使電子和離子能夠良好地進入Si負極。在鋰化過程中,Si 轉變為 Li x Si 并經歷非晶化。同時,SE 被電化學還原。理論上,最終分解產物為Li 2 S、Li 3 P和LiCl,并在界面處形成了一層薄的鈍化層。盡管該層在一定程度上會提高了離子和電子傳導的阻抗,但是它也抑制了其余部分的SE的分解。脫鋰后,納米硅變為非晶態,SE和碳的存在使復合硅負極具有較少的空隙形成。Si-SE 負極中唯一的電子傳導通路是沿著Si本身。考慮到初始Si的表面通常覆蓋有低電導率SiO 2薄層,因此Si-SE中的電子傳導相對緩慢。Si-SE負極中的Si在脫鋰后非晶化,由于巨大的體積變化,Si-SE 負極中會產生空隙。對于純Si 負極,其在鋰化和脫鋰過程中具有良好的化學穩定性。然而作為唯一相,電子傳導和離子擴散都依賴于Si,其傳導率是遠低于碳和SE的,因此反應動力學受到限制。同時,由于沒有緩沖成分,整個負極經歷了巨大的體積變化,完全脫鋰后,Si負極中形成了許多尺寸交大的孔洞和空隙。
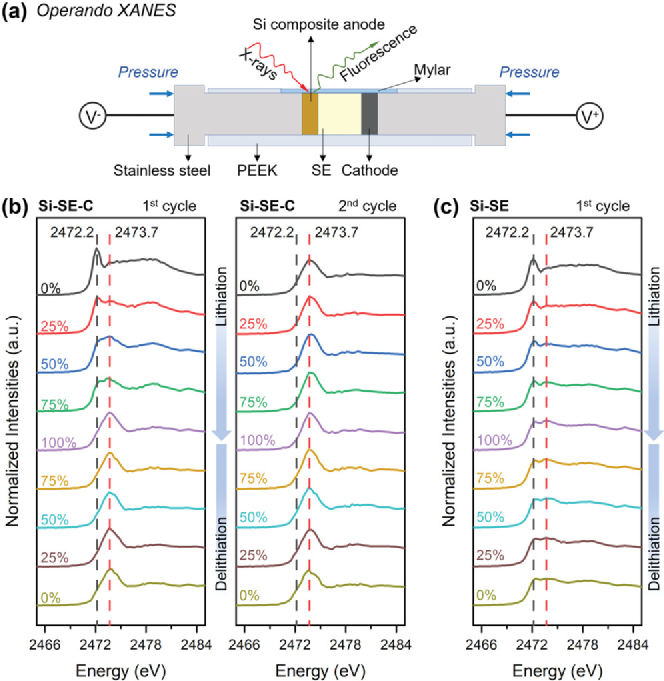
圖2是對于納米硅復合負極電化學穩定性的原位研究。圖2 b) 是Si-SE-C 復合負極在前兩個循環中不同 SoC 和 DoD 下的硫K邊XANES光譜。可以觀察到2472.2 eV處的峰逐漸消退,而2473.7 eV處的峰逐漸上升,并在最高鋰化態占據主導地位。光譜變化表明 SE 的結構在 Si 鋰化過程中發生了變化。理論上,SE 降解后的最終產物是 Li2S、Li3P 和 LiCl。在第二個循環中,光譜在鋰化和脫鋰過程中沒有明顯變化。這表明SE分解主要發生在第一個循環中,SE和其分解產物的混合物保持了長期的化學穩定。電池測試后,我們測量了其他位置的硫K邊 XANES 光譜,發現許多未反應的SE。這表明只有附著在 Si 和碳上的SE發生了降解。圖2 c) 是Si-SE 復合負極在第一個循環中不同 SoC 和 DoD 下的硫K邊XANES光譜。在鋰化過程中,峰的演變類似于 Si-SE-C 復合材料中的峰, 但是2472.2 eV 的峰即使在完全鋰化狀態下也不會消失。這表明 Si-SE 中 SE 的降解不像 Si-SE-C 中那么嚴重。在脫鋰過程中,所有峰都保持不變,證明SE的分解是不可逆的,且分解產物相對穩定。
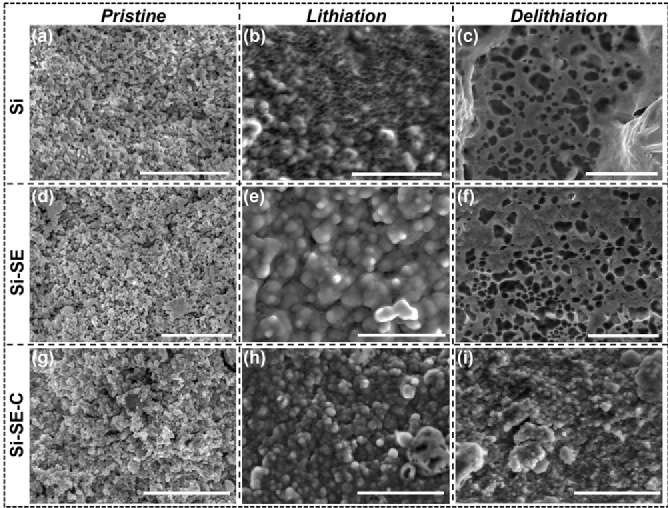
圖3 是使用SEM 對不同納米硅負極結構演變進行的研究。Si 負極a) 原始狀態、b) 鋰化后和 c) 脫鋰后的橫截面 SEM 圖像。Si-SE 復合負極d) 原始狀態、e) 鋰化后和 f) 脫鋰后的橫截面 SEM 圖像。Si-SE-C 復合負極在 g) 原始狀態、h) 鋰化后和 i) 脫鋰后的橫截面 SEM 圖像。比例尺為 2.0 μm。在鋰化過程中,Li 逐漸與 Si 形成 LixSi 合金,并伴隨著體積膨脹,電極中的納米硅粒子逐漸變為泥狀的無定形形態。另外在測試期間框架對電池材料施加了150 MPa 的軸向壓力。由于非晶 Li x Si 的機械強度低于純 Si,因此在外部壓力和體積膨脹產生的內部應力下,Li x Si的變形使整個電極致密化。脫鋰后,從 Li x Si到 Si的過程導致了巨大的體積收縮。在Si陽極中, 與原始狀態下相比(圖 3a), 脫鋰后形成了大量的尺寸較大的孔隙 (圖 3c )。相比之下,Si–SE–C(圖 3i) 在脫鋰后有最穩定的結構表現。Si-SE-C 在脫鋰后表現出更均勻和更致密的形態。這是因為分散在 Li x Si聚集體中的 SE 和 C 顆粒促進了均勻的脫鋰過程并避免了大孔的生長。因此,將 Si 與 SE 和碳復合有利于電極結構穩定性,減少孔隙生成,并保持良好的電子傳導性和離子擴散性。

圖4為使用XnT技術重建的各種納米硅負極在不同狀態下的3D結構。在初始的Si 負極中(圖 4a),立方體中只有硅納米粒子和孔隙。X 射線衰減較低的區域代表樣品中的孔隙,并用黃色標記。與 SEM 相似,原始硅陽極顯示出高度多孔的結構,計算孔隙率為 26.17%。這些孔隙是 Si 納米粒子之間的空隙。鋰化后,電極變得更加致密。Si 納米顆粒被具有更大尺寸的無定形Li x Si代替,孔隙率大大降低至 6.24%。完全脫鋰后孔隙率增加到 15.99%。
孔隙率略低于原始硅陽極,這是因為由于變形,立方體中壓縮了更多的硅。然而在脫鋰硅負極中存在一些大至微米級的隨機分布的空洞和孔隙。這些孔是合金化/脫合金化過程中 Si 巨大體積膨脹/收縮的結果,表明 Si 陽極具有不穩定的結構。由于復雜的成分,Si-SE-C 顯示出不同的結構(圖 4g)。碳幾乎不吸收x射線,因此在 XnT 中顯示出非常深的灰色。由于對比度較低,無法區分出炭中的孔隙。因此,碳內部的孔和碳被組合標記為橙色。鋰化后,產生了大塊的深灰色區域,占據了電極的很大一部分。考慮到 Si 鋰化后降低了相同子體積內的平均電子密度,從而導致了較低的X 射線衰減,因此這些區域主要歸屬于鋰化程度較高的LixSi。孔、碳、和高度鋰化的LixSi結合在一起在圖4h中用紅色標記。脫鋰后,Si-SE-C負極非常均勻(圖 4i),且產生的孔徑遠小于脫鋰的Si負極。同時,與Si-SE負極相比,孔聚集較少。因此,添加 SE和C 有利于 Si 負極的結構穩定性。
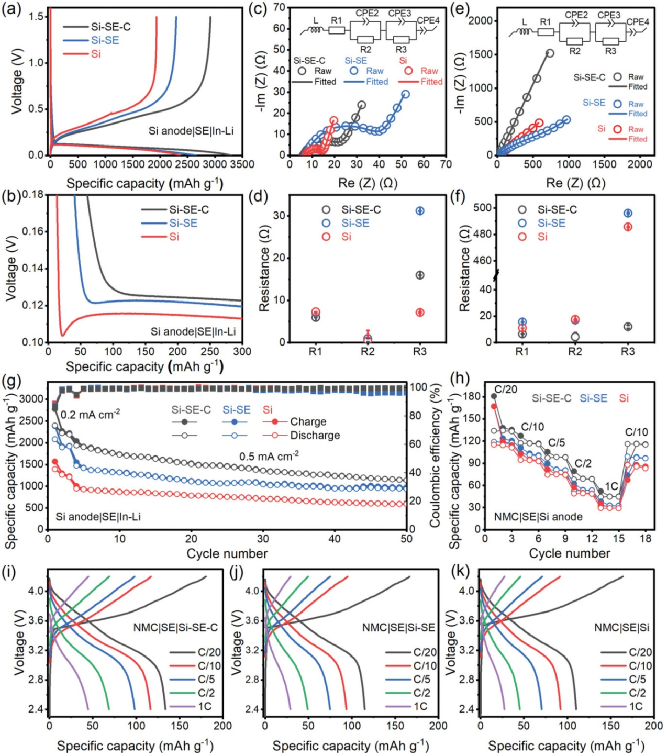
圖5為Si-SE-C、Si-SE 和 Si在半電池和全電池中的電化學性能。Si-SE-C 復合負極表現出了更優異的性能。圖 5a顯示了初始循環中所有三個電極在 0.1 mA cm -2電流密度下的恒電流充電和放電曲線。Si–SE–C 提供最高的放電/充電容量 3288/2917 mAh g ?1,初始庫侖效率 (ICE) 為 88.7%。相比之下,Si-SE 的放電/充電容量相對較低,為 2653/2291 mAh g -1,ICE為 86.4%。純硅負極顯示出最低的放電/充電容量 2353/1935 mAh g -1,ICE 為 82.2%。Si-SE-C 負極的最高容量和 ICE 表明將 Si 與 SE 和碳復合有利于獲得最佳的 Si 利用率。圖 5b放大了初始鋰化過程中的充電/放電曲線。純Si電極存在明顯的鋰化過電位,而Si-SE-C和Si-SE直接表現出平坦的鋰化平臺。這種過電位主要是由硅中緩慢的離子擴散和電子轉移引起的。我們進一步研究了三種硅負極在全電池中的電化學性能。Si-SE-C 電池提供最高的平均放電容量和倍率性能。同時,Si-SE-C 的全電池在前三個循環中表現出了最佳穩定性。這些結果表明,SE 的分解對硅負極的影響可以忽略不計,結構穩定性是更重要的因素。

圖6為使用通過化學-彈性-塑性模型(chemo-elasto-plastic mode)對不同納米硅負極在鋰化和脫鋰過程的大形變結構演化研究。Si-SE-C 表現出顯著較低的塑性變形和較少的Von Mises 應力產生。這是因為與 Si 相比,SE 和 C 的硬度要小得多,在 Si-SE-C 復合負極中充當了應力松弛介質。
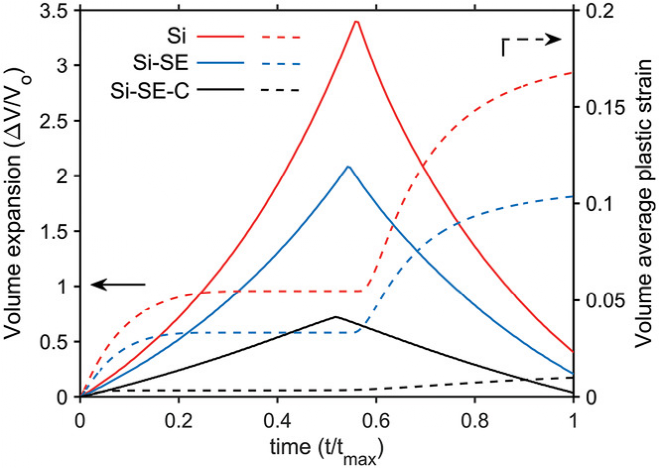
圖7為鋰化何脫鋰過程中不同負極材料的體積膨脹和收縮以及平均體積塑性應變。Si-SE-C 負極表現出了最低的體積膨脹和平均體積塑性應變。從上述模擬中獲得的結果說明,將 SE 和 C 添加到 Si 中可以成為提高 Si 負極材料力學穩定性的可行方案。
審核編輯 :李倩
-
鋰離子電池
+關注
關注
85文章
3319瀏覽量
78696 -
電化學
+關注
關注
1文章
330瀏覽量
20893 -
負極材料
+關注
關注
12文章
177瀏覽量
14749
原文標題:硫化物基全固態電池中納米硅復合負極的電化學和力學機理研究
文章出處:【微信號:Recycle-Li-Battery,微信公眾號:鋰電聯盟會長】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
新型銅互連方法—電化學機械拋光技術研究進展
蘋果的新專利--全固態電池
電化學原理介紹和分析方法
鋰硫電池電化學循環過程及其正極反應機理的研究進展介紹

電化學阻抗譜的設計基礎
原位核磁共振研究硫化物基全固態鋰電池失效機理
通過原位固體核磁共振理解硫化物基全固態鋰金屬電池的失效過程
AEM綜述:硫化物基固態鋰電池的先進表征技術
鋰離子電池負極衰減機理研究進展
全固態電池的單片100%硅片負極在室溫下實現高面積容量

清華大學:自由空間對硫化物固態電解質表面及內部裂紋處鋰沉積行為的影響





 工商網監
工商網監

評論