") 打開穩(wěn)定低溫鋰金屬負極的極化和可逆性限制
打開穩(wěn)定低溫鋰金屬負極的極化和可逆性限制
研究背景
近年來,高濃縮電解液(HCE)、局部高濃縮電解質(zhì)(LHCE)、和弱溶劑化電解液(WSE)的新設(shè)計概念將鋰金屬負極的循環(huán)可逆性帶入了一個新時代,其中的核心謎團是陰離子衍生的SEI。這種獨特的SEI化學(xué)具有更有利的特性,包括空間均勻性、化學(xué)惰性和機械魯棒性,以減輕SEI本身的損傷和生長。此外,在陰離子衍生的SEI下,Li沉積方式可以從針狀形態(tài)定制成更均勻的顆粒形狀大大減少了電隔離的死鋰的數(shù)量。這兩個方面的共同貢獻有助于大大提高工作的鋰金屬負極的循環(huán)可逆性。
雖然這些電解質(zhì)設(shè)計的新進展明確地提高了鋰金屬負極的室溫性能提高到一個全新的水平,但工作電池實際上受到不同溫度條件的影響。深入了解溫度依賴的Li沉積/溶解行為,以及揭示溫度依賴的庫侖效率的關(guān)鍵決定因素,具有重要意義。在這種背景下,最近的一些研究已經(jīng)表明,提高工作溫度在一個合理的范圍內(nèi)可以促進鋰金屬負極的性能,通過界面和動力學(xué)的好處,與之形成鮮明對比的是,鋰金屬負極的零下溫度性能遠不令人滿意,其主要瓶頸在于工作溫度降低時極化程度較大,可逆性較差(圖1)。
實際上,鋰沉積的完整圖像包括體離子輸運、界面鋰離子(Li+)脫溶、Li+在SEI中的擴散和電子轉(zhuǎn)移。如Vogel–Tammann–Fulcher (VTF)方程和阿倫尼烏斯方程所述,這四種過程的動力學(xué)速率本質(zhì)上都依賴于溫度,而最電阻步驟(速率決定步驟)仍存在爭議。此外,工作溫度的降低通常伴隨著鋰沉積/溶解的可逆性顯著降低,這也是一些表現(xiàn)出優(yōu)越的室溫性能的電解質(zhì)的情況。揭示低溫鋰沉積過程中的動力速率決定因素,闡明鋰金屬負極的極化問題和可逆性問題之間的相互作用,是構(gòu)建穩(wěn)定低溫鋰金屬電池的首要步驟。
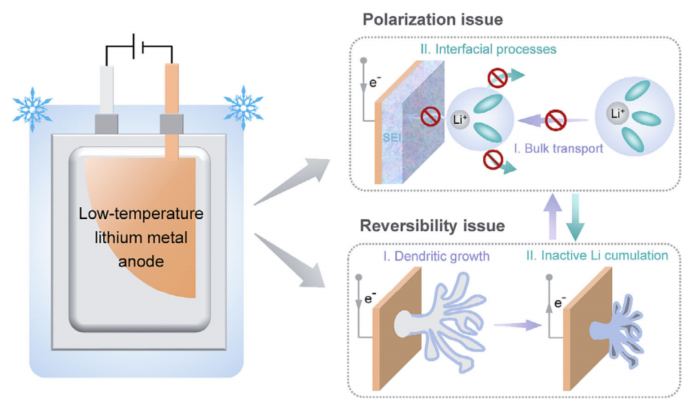
圖 1 穩(wěn)定低溫金屬鋰負極的主要瓶頸,即極化問題、可逆性問題以及這兩個因素如何相互作用的示意圖。
成果簡介
近日,北京理工大學(xué)黃佳琦教授對鋰沉積過程中的過電位屬性進行了分解,并建立了動力學(xué)過電位支配、SEI化學(xué)的動態(tài)演化和相應(yīng)的Li可逆性之間的相互作用。通過采用典型的LHCE作為模型系統(tǒng),揭示了離子濃度梯度發(fā)揮壓倒性的作用在極化正極鋰沉積過程在零下溫度下,這不僅直接限制工作電池的功率輸出,而且嚴(yán)重降低鋰沉積均勻性的有限的陰離子分解動態(tài)SEI形成。受研究結(jié)果的啟發(fā),將低溫電解質(zhì)的設(shè)計合理解耦,同時抑制工作過電位,為SEI的動態(tài)形成提供可持續(xù)的協(xié)調(diào)陰離子供應(yīng)。因此,在1.0 mA cm-2下,-20°C的Li沉積/溶解CE明顯從95.93%提高到98.40%,動力學(xué)過電位降低,在實際的Li|LiNi0.5Co0.2Mn0.3O2(NCM523)電池中進一步證明壽命延長了3倍以上。
研究亮點
(1) 探究了在不同溫度不通電流密度下的局部高濃電解液體系里的鋰沉積行為。
(2) 將鋰金屬沉積過電位分解,通過時間分辨的弛豫測量分析出濃度過電位、界面反應(yīng)過電位、歐姆過電位,并探究收溫度、電流密度對極化電位的影響。
(3) 通過核磁定量的探究在不用的電流密度下的電解質(zhì)中DME與FSI-的消耗,在較大的Li+濃度梯度下,Li+-陰離子相互作用減弱,不利于陰離子衍生的SEI形成。
(4) 合理的設(shè)計出一款低過電位和高鋰可逆性的低溫電解質(zhì)的設(shè)計策略,引入了微量(0.05M)的強配位硝酸鋰(LiNO3),以補償Li+-陰離子的聚集損失。
圖文導(dǎo)讀
局部高濃電解液中的低溫鋰沉積行為
1.0 M LiFSI溶解于DME/TTE電解質(zhì)標(biāo)記為基線LHCE,此電解液被廣泛報道但低溫性能仍有待研究。首先,系統(tǒng)地比較了Li | Cu電池在25和-20°C時的Li沉積行為。如圖2a、b所示,平均CEs小電流密度0.2 mA cm-2時比較類似,而當(dāng)電流密度提升到1.0 mA cm-2平均CE的差異變大。在25°C時Li+可逆沉積/溶解的庫侖效率為99.12%,-20°C的庫侖效率降低至95.93%,這不利于長壽命可充電電池。圖2c、d進一步記錄了不同溫度和電流密度下的Li沉積形態(tài),以找出不同庫侖效率的原因。
在0.2 mA cm-2的小電流密度下,Li沉積物在25和-20°C下均呈現(xiàn)出良好的大顆粒狀形態(tài)(圖2c),這與它們在Li | Cu電池中獲得的庫侖效率很一致(圖2a)。當(dāng)電流密度增加到1.0 mA cm-2,即使在25°C下可以保持顆粒狀鋰沉積形態(tài),當(dāng)工作溫度降低到-20°C時,它變成了具有高縱橫比針狀形態(tài)的高度樹突狀結(jié)構(gòu)(圖2d),Li沉積行為的急劇轉(zhuǎn)變導(dǎo)致可逆性明顯下降。
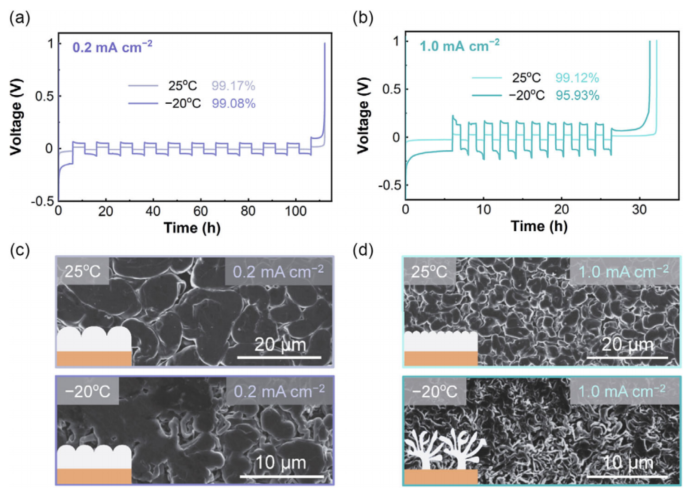
圖 2 LHCE電解液中不同溫度和電流密度下的鋰沉積行為,包括不同溫度下Li|Cu半電池在(a)0.2mA cm-2和(b)1.0 mA cm-2電流密度下的平均庫侖效率,和不同溫度下Li|Cu半電池在(c)0.2mA cm-2和(d)1.0 mA cm-2電流密度下的的沉積形貌。在(a,b)循環(huán)和(c,d)沉積中的總?cè)萘繛?.0 mAh cm-2。
在研究界廣泛承認(rèn)的是,Li的沉積行為主要由SEI控制,而SEI的形成過程直接由Li+的溶劑化結(jié)構(gòu)決定。基于這一觀點,通過分子動力學(xué)(MD)模擬,直觀地研究了與溫度相關(guān)的Li+溶劑化結(jié)構(gòu)。模擬結(jié)果如圖3a、b,降低溫度提高了陰離子-陽離子交互強度,這可以通過在Li+的初級溶劑化殼中Li+和FSI-之間的Li-O相互作用更強的峰驗證,因此將溫度從25降低到-20°C后,F(xiàn)SI-的平均配位數(shù)增強(從2.61到2.76)。此外,特定Li+配位態(tài)的微觀統(tǒng)計顯示,DME分子單獨溶劑化的Li+百分比(3-0-0)從5.0%顯著下降到0.8%(圖3c,d)。
根據(jù)已建立的理論,一個更強的陰離子-陽離子配位將促進更多的陰離子分解,以促進陰離子衍生的SEI的形成。為了證實這一點,我們進行了電化學(xué)阻抗譜(EIS)表征,以探測在不同溫度下長期靜置以形成靜態(tài)SEI的對稱Li電池的界面電阻,-20°C時的電池電阻低于25°C時的電阻,而由于SEI富無機性質(zhì),兩種SEI具有相似的離子激活能(Ea)。以上觀察明顯強調(diào),靜態(tài)Li+溶劑化結(jié)構(gòu)的變化不應(yīng)該是決定鋰沉積方式轉(zhuǎn)變的主要原因,而是更可能是由于實際工作的Li負極的動態(tài)相關(guān)界面行為導(dǎo)致的。
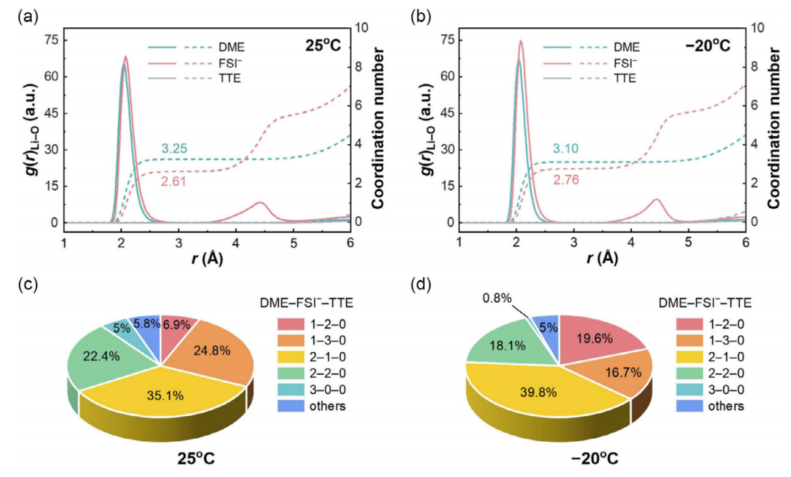
圖 3 通過MD模擬了具有溫度依賴性的Li+溶劑化結(jié)構(gòu)。根據(jù)(a) 25°C和(b)-20°C,通過MD模擬軌跡計算的Li -ODME,Li-OFSI- 和Li-OTTE對的徑向分布函數(shù)(實線)和配位數(shù)圖(虛線),(c,d)是不同Li+配位結(jié)構(gòu)的相應(yīng)統(tǒng)計。
低溫下鋰沉積的動態(tài)研究
由上述演繹,一個時間分辨瞬態(tài)弛豫測量來分析動態(tài)過電位屬性在低溫鋰金屬沉積(圖4),其中一個簡短的弛豫步驟(10.0s)是間歇地引入探頭連續(xù)恒流鋰沉積過程中區(qū)分濃度過電位(ηconc)和iR下降(圖4a、b)。重要的是,iR下降被定義為弛豫步驟前的最終電位值與指定靜置時間后的弛豫電位值之間的突然電位下降。合理選擇界面電荷轉(zhuǎn)移過程的弛豫時間為時間常數(shù)(τ)的5倍,以確保界面反應(yīng)過電位(ηreac)完全釋放,使其合并到iR下降中。然后,考慮到ηohm通常不受鋰沉積深度的影響,總iR可以被分解為歐姆過電位(ηohm)和ηreac。在圖4c、d中可以清晰的分解。結(jié)果似乎與之前的認(rèn)識相反,即界面過程(Li+通過SEI或Li+脫溶態(tài))是低溫負極的動力學(xué)速率決定步驟;這里發(fā)現(xiàn),在-20°C的Li沉積中,在0.2和1.0 mA cm-2下,ηconc對總過電位的貢獻最大(圖4c、d)。
值得注意的是,當(dāng)將Li沉積溫度升高到25°C時,過電位的支配地位轉(zhuǎn)移到了ηreac,這可以從溫度從-20°C(0.21 mS cm-1)提高到25°C(0.70 mS cm-1)時離子電導(dǎo)率的顯著變化中解釋。此外,我們發(fā)現(xiàn),無論電流密度(圖4c,d)和溫度(如何,ηreac都隨著Li沉積容量的增加而降低。其主要原因是隨著鋰沉積深度的增加,活性表面積逐漸增大,從而降低了界面上電化學(xué)反應(yīng)的電阻和過電位。進一步進行了COMSOL模擬,從理論上考察了低溫鋰沉積條件下離子濃度的演化(圖4e、f)。當(dāng)電流密度從0.2 mA cm-2,增加到1.0 mA cm-2時,Li+載流子的消耗和補充明顯失衡,導(dǎo)致連續(xù)沉積鋰后負極表面的離子濃度降低了近一半(圖4f)。
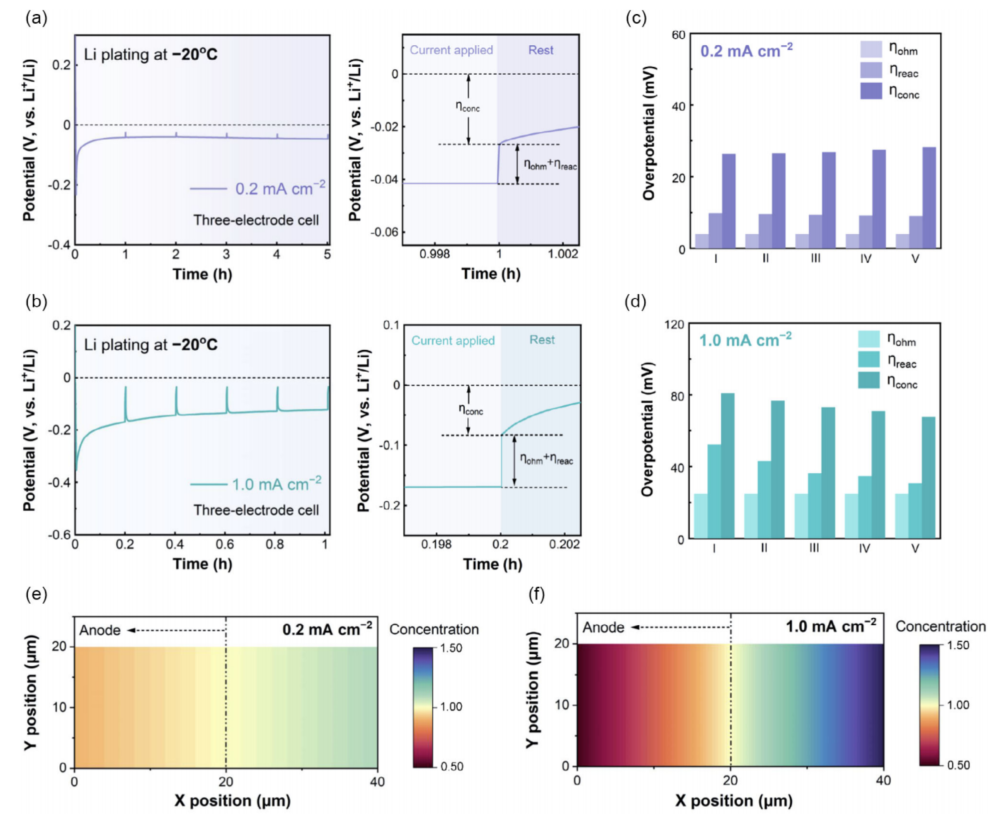
圖 4 在(a)0.2 mA cm-2和(b)1.0 mA cm-2下,三電極時間分辨瞬態(tài)弛豫測量對低溫鋰沉積的過電位屬性進行分解在(c)和(d)中得出了不同的的ηohm、ηCT和ηconc在(e) 0.2 mA cm-2和(f)1.0 mA cm-2下沉積1.0mAh cm-2后,電解液中離子濃度梯度的COMSOL模擬結(jié)果。
一方面,如Sandy的時間模型所述,Li+濃度的耗盡有可能引發(fā)Li樹突的生長。另一方面,局部Li+濃度的演化應(yīng)該會強烈地改變原來的Li+陰離子相互作用,而在Li+濃度動態(tài)降低的情況下,Li+陰離子相互作用逐漸減弱,預(yù)計將極大地影響SEI的動態(tài)形成過程。為了證實后一點,我們通過核磁共振(NMR)技術(shù)定量地研究了陰離子和溶劑對SEI漸進形成的動態(tài)消耗。本研究有意利用無SEI、結(jié)構(gòu)穩(wěn)定的Li7Ti5O12電極為Cu上的鋰沉積提供Li源,以必然排除對電極的電解質(zhì)消耗(圖5a)。分別在19F和1H NMR下(圖5b)測定了Cu | Li7Ti5O12電池10個循環(huán)后的電解質(zhì)。通過追蹤FSI-(19F譜)和DME(1H譜)相對于內(nèi)參試劑的峰值強度演化,可以很容易地定量鹽和溶劑的動態(tài)消耗。如圖5c所計算,在0.2 mA cm-2的電流密度下的FSI-消耗(9.5%)比DME(7.2%)更顯著,而在1.0 mA cm-2下選擇性消耗DME,因為DME的損失(11.4%)顯著高于FSI-(3.8%)。
通過x射線光電子光譜(XPS)進一步檢測了在0.2和1.0 mA cm-2下Li沉積后的SEI化學(xué)性質(zhì)。如圖5d、e所示,SEI形成于在0.2 mA cm-2富含F(xiàn)SI-的分解產(chǎn)物L(fēng)iF、Li2O, Li3Nand Li2S。然而,當(dāng)電流密度增加到1.0 mA cm-2時,這些陰離子衍生的SEI物種的數(shù)量明顯減少上述結(jié)果生動地表明,動態(tài)電解質(zhì)消耗SEI形成反應(yīng)和由此產(chǎn)生的SEI可以調(diào)控的實時Li+-陰離子交互界面,在較大的Li+濃度梯度下Li+-陰離子結(jié)合強度減弱,不利于陰離子衍生的SEI的形成。
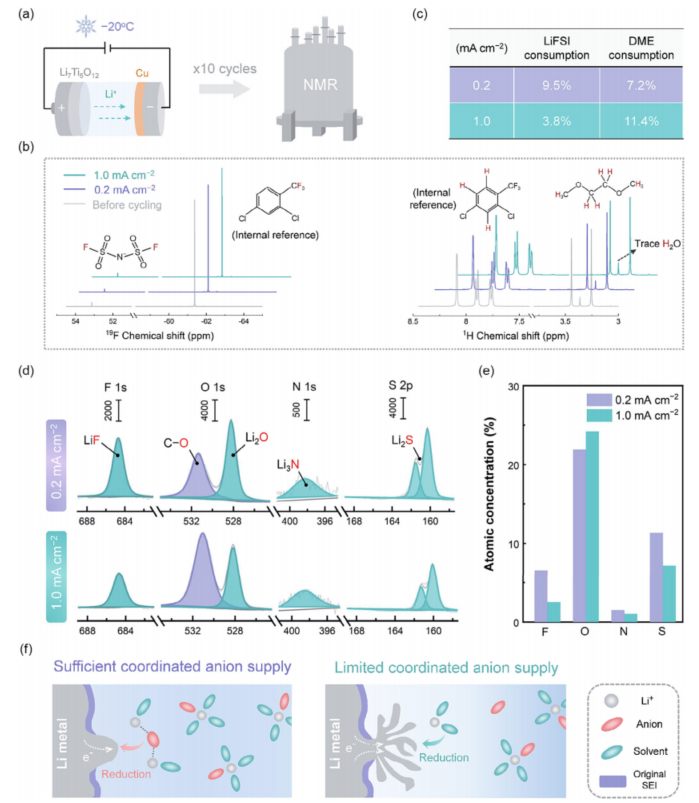
圖 5 通過核磁共振技術(shù)測定陰離子和溶劑的消耗,包括(a)表征程序的示意圖,(b)未循環(huán)和循環(huán)電解質(zhì)的19F和1 H NMR譜,以及(c)計算出的FSI-和DME的消耗比。在XPS下對-20°C的Li沉積物進行SEI化學(xué)分析,包括(d)F 1s、O 1s、N 1s和S 2p光譜,以及(e)相應(yīng)的原子濃度。這些數(shù)據(jù)是在120 s的濺射后獲得的。(f)在協(xié)調(diào)陰離子供應(yīng)充足和有限的情況下,動態(tài)SEI修復(fù)過程的示意圖,這分別導(dǎo)致顆粒狀和樹突狀Li沉積。
為了進一步證實Sandy的時間模型或動態(tài)界面化學(xué)模型是決定Li沉積和電池可逆性的決定因素,我們設(shè)計了一個通過調(diào)整DME的體積比例到TTE的對照實驗。當(dāng)提高體積/TTE從1/2到2/1,離子電導(dǎo)率-20°C可以提高38.1%到0.29 mS cm-1。受益于增強鹽解離,有利于明顯的緩解極化鋰沉積/剝離。然而,這種加速Li+擴散的好處并沒有導(dǎo)致對Li可逆性的任何預(yù)期的增強。相反,平均庫侖效率甚至從接近96%下降到95.31%,這可以歸因于一旦降低了體積電解質(zhì)中有效的Li+濃度(DME中的Li+濃度),Li+-陰離子相互作用不足,導(dǎo)致陰離子還原。
在這里,我們可以安全地得出結(jié)論,實時Li+濃度依賴的Li+-陰離子相互作用和相應(yīng)的動態(tài)SEI形成途徑是決定Li沉積行為,從而決定電池低溫下沉積行為可逆性的關(guān)鍵因素。當(dāng)配位陰離子供應(yīng)充足時,裂解的SEI可以通過陰離子的分解迅速修復(fù)。因此,可以保留顆粒狀鋰的沉積方式。然而,當(dāng)產(chǎn)生較大的離子濃度梯度時,Li+-陰離子的相互作用減弱,配位陰離子的數(shù)量受到限制,難以維持SEI的持續(xù)修復(fù)。在這種情況下,陰離子衍生的SEI化學(xué)逐漸變成溶劑衍生的化學(xué),這將鋰沉積形態(tài)轉(zhuǎn)變?yōu)獒槧顦渫粻钅J健?/p>
低溫工作調(diào)節(jié)鋰金屬負極的電極液的合理設(shè)計
根據(jù)以上討論,我們進一步探討了同時針對低過電位和高鋰離子可逆性的低溫電解液的設(shè)計策略(圖6a)。本質(zhì)上,濃度過電位的降低需要較低的Li+/DME比值來促進鋰鹽的解離。相反,維持一個強大的Li+-陰離子相互作用對于有利的陰離子衍生的SEI形成需要一個足夠高的Li+/DME比值。為了克服這種權(quán)衡困境,我們提出在電解質(zhì)設(shè)計中分離快速的Li+轉(zhuǎn)運的功能和持續(xù)的協(xié)調(diào)陰離子供應(yīng),在電解質(zhì)設(shè)計中動態(tài)SEI形成。我們將DME/TTE(1/2 by vol.)中LiFSI鹽的濃度略微降低到0.8M,這促進了更高的離子電導(dǎo)率(0.31 mS cm-1),從而顯著降低了電池的極化。此外,還引入了微量(0.05 M)的強配位硝酸鋰(LiNO3)來補償Li+-陰離子聚集的損失。值得注意的是,增強的LiNO3(0.10 M)將極大地影響電解質(zhì)的低溫離子電導(dǎo)率,甚至低于LHCE電解液。這與文獻中觀察到的傳統(tǒng)含LiNO3的電解質(zhì)的低溫性能較差相一致。
不同的是,本研究中開發(fā)的先進電解質(zhì)在1.0 mA cm-2下,Li|Cu電池的平均CE提高到98.40%,并降低了鋰沉積過電位(圖6b)。進一步采用滴定氣相色譜(TGC)分析了循環(huán)電池的不可逆性來源(圖6c)。結(jié)果表明,高級電解質(zhì)中產(chǎn)生的死Li量僅為LHCE電解質(zhì)的五分之一,這與它們明顯的Li沉積密切相關(guān)。在LiNO3衍生的SEI化學(xué)條件下,球形的Li形態(tài)是一種典型的Li沉積模式。此外,結(jié)果表明,SEI- Li+容量損失相當(dāng)?shù)碾娊赓|(zhì),這可以解釋為球形鋰的表面積降低,LiNO3的高反應(yīng)性可能會加重單位表面積的鋰消耗。這兩個因素的共同貢獻最終導(dǎo)致了SEI- Li+容量的變化可以忽略不計。
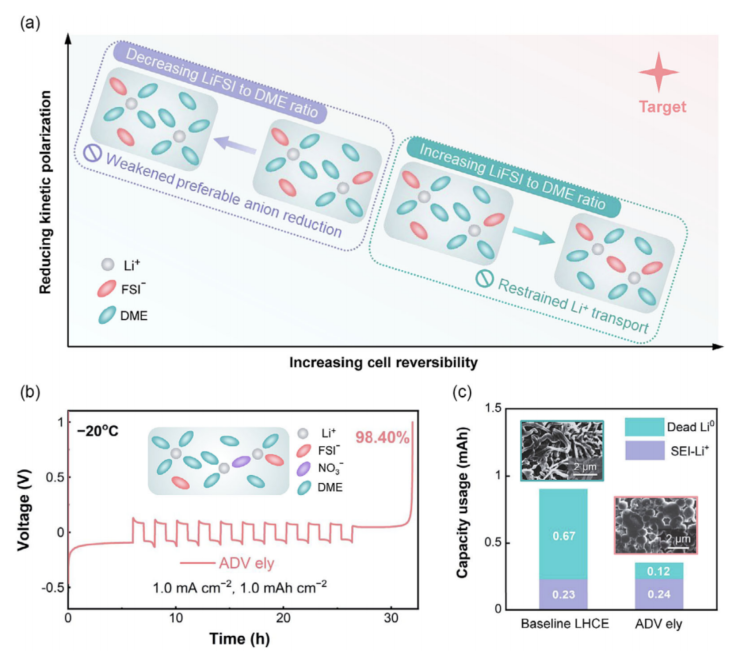
圖 6 (a)低溫電解質(zhì)設(shè)計的示意圖,同時針對低過電位和高鋰可逆性,其中僅調(diào)節(jié)鋰鹽濃度是無法實現(xiàn)的。(b)利用先進的電解質(zhì)測定Li | Cu半電池的平均CE。該插圖說明了在優(yōu)化電解質(zhì)中形成的典型溶劑化結(jié)構(gòu)。(c)在1.0 mA cm-2下進行10個循環(huán)后的非活性Li組成的TGC結(jié)果,插圖是LHCE和ADV電解質(zhì)中各自的Li沉積形態(tài)。
Li | NCM523電采用有限的Li源(50 μm)、高負載正極(≈3.3mAh cm-2)和有限的電解液(7.0 μL mAh-1)用于低溫循環(huán)(圖7)。低溫電池在初始循環(huán)中需要更長的激活期,這可能是由于電極潤濕和SEI/CEI積累的動力學(xué)緩慢。在0.1 C的低速率下,LHCE的電池可以循環(huán)60個周期,其容量保持率為80%(圖7a)。相比之下,改善后的電解質(zhì)在很大程度上增強了Li | NCM523電池的循環(huán)穩(wěn)定性,在200個循環(huán)中可以觀察到容量的衰減可以忽略不計(圖7a)。進一步將電流速率提高到0.25 C,兩個電池的性能下降(圖7b)。盡管如此,與對比樣的短壽命相比(只有30個周期),使用優(yōu)化的電解質(zhì)的電池的使用壽命仍然延長了近3倍(圖7b)。這種容量的衰減可以直接反映在電壓曲線上(圖7c,d),其中極化增強被確定為電池持續(xù)容量損失的根源。
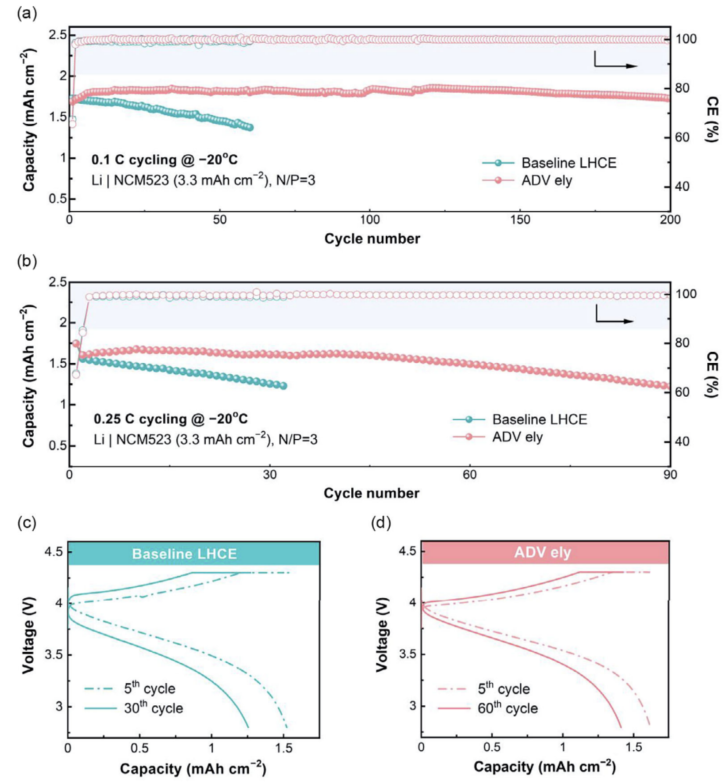
圖 7 實際的Li | NCM523電池在(a)0.1 C和(b) 0.25 C下循環(huán)的低溫(-20°C)性能,以及使用c) LHCE和d)ADV電解質(zhì)在0.25 C下記錄的電壓曲線。
有趣的是,如果在-20°C下無法循環(huán)的電池重新進行室溫循環(huán),就可以消除如此大的過電位,容量損失幾乎完全恢復(fù)。從這種非永久性容量損失現(xiàn)象可以推斷,逐漸積累的非活性Li層內(nèi)不斷降解的電解質(zhì)潤濕應(yīng)該是低溫Li | NCM523電池極化增強的原因。這種由死鋰和裂解的SEI組成的多孔層,一旦溫度升高,就可以被電解質(zhì)重新滲透,有助于減輕大極化和恢復(fù)電池容量。這說明低溫電池的工作極化對非活性鋰的產(chǎn)生更加敏感,這與電解質(zhì)的物理性質(zhì)(粘度、離子電導(dǎo)率等)的變化有關(guān)。
總結(jié)與展望
綜上所述,本工作仔細研究了低溫鋰沉積的過電位屬性,并以LHCE為模型平臺,建立了過電位支配、SEI化學(xué)的動態(tài)演化和相應(yīng)的鋰?yán)眯手g的相關(guān)性。與常識不同的是,我們發(fā)現(xiàn)在零下工作溫度下,濃度過電位主導(dǎo)了整個鋰沉積過電位,這一方面限制了動力學(xué)性能,另一方面通過限制動態(tài)SEI形成過程中的陰離子分解,降低了鋰沉積的均勻性。通過復(fù)雜地設(shè)計電解質(zhì)化學(xué),可以同時減少工作過電位和持續(xù)提供配位陰離子以形成動態(tài)SEI。因此,在-20°C下,Li | NCM523電池的CE沉積/溶解效率從95.93%明顯提高,實際Li | NCM523電池提高到98.40%,壽命進一步延長了3倍以上。本工作揭示了低溫工作鋰金屬負極極化和可逆性的實際因素,為實用鋰金屬電池在零下溫度下穩(wěn)定高效循環(huán)提供了新的設(shè)計原則。
審核編輯:劉清
-
電解液
+關(guān)注
關(guān)注
10文章
848瀏覽量
23095 -
FSI
+關(guān)注
關(guān)注
0文章
4瀏覽量
5892 -
WSE
+關(guān)注
關(guān)注
0文章
6瀏覽量
7642 -
鋰金屬電池
+關(guān)注
關(guān)注
0文章
136瀏覽量
4311
原文標(biāo)題:打開穩(wěn)定低溫鋰金屬負極的極化和可逆性限制
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關(guān)注!文章轉(zhuǎn)載請注明出處。
發(fā)布評論請先 登錄
相關(guān)推薦
多功能高熵合金納米層實現(xiàn)長壽命無負極鈉金屬電池

離子液體添加劑用于高壓無負極鋰金屬電池

通過電荷分離型共價有機框架實現(xiàn)對鋰金屬電池固態(tài)電解質(zhì)界面的精準(zhǔn)調(diào)控
Nat. Commun.:新型固態(tài)電池負極,高穩(wěn)定、快充

全固態(tài)鋰金屬電池的鋰陽極夾層設(shè)計
真空蒸發(fā)電鍍可用于鋰金屬電池的≤10μm超薄鋰箔

最新Nature Energy開發(fā)新型稀釋劑助推鋰金屬電池實用化!

用于延長高壓高Ni三元鋰金屬電池壽命的無氟醚基電解液

聚焦無枝晶生長的負極設(shè)計
具有分級脫嵌鋰機制的Li多相合金負極

全固態(tài)鋰金屬電池負極界面設(shè)計


通過金屬負極/LPSCl界面調(diào)控實現(xiàn)超穩(wěn)定全固態(tài)鋰金屬電池

人工界面修飾助力高性能鋰金屬電池的最新研究進展與展望!





 工商網(wǎng)監(jiān)
工商網(wǎng)監(jiān)
評論