

背景介紹
可充電鎂金屬電池由于鎂的高豐度、高容量和良好的安全性而受到了極大的關注。在其電化學還原/氧化過程中,Mg經歷兩次電子轉移,可實現3833mAhmL-1的體積容量;Mg不容易形成Mg枝晶,這使得Mg金屬負極比其他金屬負極(如Li、Na和K)更安全。然而,表面鈍化和緩慢的電化學動力學顯著限制了鎂金屬負極的應用。鎂金屬負極表面形成的界面層通常是Mg2+絕緣的。形成活性[Mg2Cl3]+二聚體并同時避免表面鈍化層的可行方法是將Cl引入電解質中。然而含Cl電解質的電化學窗口很窄,與商業集流體不兼容。因此,開發用于鎂金屬電池的無Cl電解質是一項重要的任務。
最近,引入了高熵概念來調節電解質的物理和化學性質,從而提高金屬負極的電化學性能。引入多個組分以增加構型熵對自由能的貢獻是抑制熵降低行為的有效方法,例如從混合溶液中分離一個組分和聚集兩個或更多組分,大大提高了金屬電極的電化學性能。
成果簡介
近日,北京大學徐東升、李琦團隊與中科大焦淑紅團隊受高熵電解質潛在優勢的啟發,開發了一種多組分高熵電解質,含有(Mg(TFSI)2、(DME)、(LiOTf)、TMP(磷酸三甲酯)),以穩定鎂金屬負極。由于OTf?和TMP的強配位能力,在Mg(TFSI)2/DME基電解質中獲得了Mg2+-2DME-OTf--Li+-DME-TMP的高熵溶劑化結構。這種結構顯著減少了Mg2+-DME的相互作用,促進了Mg2+導電層在鎂金屬負極上的形成,防止了Mg金屬負極上絕緣成分的形成。該實驗中,鎂-銅電池表現出高達98%的高庫侖效率,鎂-鎂對稱電池在450小時內表現出低電壓滯后。此外,Mg||Mo6S8全電池在200次循環內表現出穩定的電化學性能。這項工作首次設計并充分表征了一種能夠成功穩定鎂金屬負極的高熵電解質。
圖文導讀 01 鎂金屬負極的電化學可逆性
DME中添加0.2MMg(TFSI)2被選為基礎電解質。通過在基礎電解質中加入LiOTf和TMP獲得兩種溶劑獲得具有兩種陽離子(Li+、Mg2+)、兩種陰離子(TFSI-、OTf-)以及兩種溶劑(DME、TMP)的高熵電解質。將Mg‖Cu電池與商用聚烯烴隔膜組裝,以評估Mg金屬負極的電化學可逆性。在最初的幾個循環中,Mg金屬負極在基礎電解質中顯示出近乎完全的不可逆性。當向基礎電解質中添加10%(v/v)TMP時,CE在第一次循環中顯著提高到58%,在40次循環后達到90%(圖1a)。有趣的是,高熵電解質中的鎂金屬負極在隨后的實驗中表現出高達98%的CE。
圖1b顯示了Mg||Mg對稱電池在0.1mA cm-2下在不同電解質中的電壓-時間曲線。采用基礎電解質的對稱電池表現出約2V的大過電位,表明負極與電解質不相容;單獨添加TMP可以有效地降低鎂金屬負極的電壓滯后,但未能延長其使用壽命;在LiOTf和TMP的共同添加下,電池在最初的幾個循環中表現出低得多的過電勢,僅為80mV,450小時后過電勢逐漸增加到580mV。因此,高熵電解質賦予電池的電化學性能,特別是其庫侖效率、壽命、過電位和氧化穩定性,與從Cl或B基電解質中獲得的結果相當。
圖1c給出了不同電解質中鎂金屬負極的CV曲線,這一結果與對稱電池的結果一致。在TMP和LiOTf的共同添加后觀察到的強電流響應證明了這些添加劑對Mg金屬負極的穩定性的協同作用。在1至0V的電壓范圍內獲得的放大CV曲線揭示了鎂金屬成核之前電解質的電化學行為(圖1d)。在TMP和LiOTf的共同添加后,出現了明顯的還原峰,表明在Mg沉積之前形成了界面層。進行了電化學阻抗譜(EIS)來研究Mg金屬負極的界面性質(圖1e)。結果有力地證明了陽極上存在界面層。在高熵電解質中形成的界面層的電阻為996Ω,證實了其Mg2+導電性。

【圖1】(a)Mg||Cu電池在Mg(TFSI)2/DME+TMP(有和無LiOTf)中的庫侖效率。所施加的電流密度為0.5mAcm-2,并且面積容量為0.1mAh cm-2。(b)0.1 mA cm-2下,不同電解質中Mg||Mg對稱電池的電壓-時間分布。(c)Mg||Cu電池在三種電解質中以10mVs-1的掃描速率的循環伏安圖。(d)Mg||Cu電池在電壓范圍為1至0V的三種電解質中的放大CV曲線
02 鎂沉積的形貌和表面化學
為了揭示添加和不添加LiOTf/TMP的Mg(TFSI)2/DME的電化學性能的差異,作者使用透射電子顯微鏡(TEM)和掃描電子顯微鏡(SEM)研究了負極上形成的Mg沉積物的結構和形態。可以看出,在Mg(TFSI)2/DME中形成了多孔界面層(420nm)(圖2a)。隨著TMP的加入,界面層變得致密,其厚度降至220nm(圖2b)。TMP和LiOTf共同加入到基電解質中導致了光滑緊湊的界面層,其厚度為140nm(圖2c),利用聚焦離子束(FIB)-SEM對鎂合金負極的形貌和結構進行了研究,在0.1mAcm-2的低電流密度和2h的長沉積時間下獲得了大的鎂合金鍍層,并對其進行了表征(圖2d和2e)。
與采用基礎電解質的沉積物形成對比的是,添加LiOTf和TMP的Mg沉積物緊密堆疊,表面層薄(圖2h、2i)。在Mg(TFSI)2/DME中形成了尺寸約為5μm的半球形Mg沉積物(圖2f)。能量色散譜(EDS)圖譜顯示,大量的Mg和O分布在Mg沉積物上,這與TEM和FIB-SEM圖像中觀察到的厚界面層一致(圖2g)。相比之下,在高熵電解質中形成的Mg沉積物光滑致密,具有明顯的晶體取向(圖2j,2k)。EDS圖譜顯示,大多數Mg沉積物和少量Au證實了其表面存在薄界面層。這種薄而緊湊的界面層具有高Mg2+電導率,有助于鎂金屬負極的高可逆性和長壽命。

【圖2】Mg沉積的TEM圖像,在(a)Mg(TFSI)2/DME、(b)Mg(TFSI)2/DME+TMP、(c)Mg(TFSI)2/DME+TMP/LiOTf;Mg在Mg(TFSI)2/DME上沉積的(d)FIB-SEM圖像,(e)相應的放大SEM圖像,以及(f)側視圖以及(g)頂視圖;Mg在Mg(TFSI)2/DME+TMP/LiOTf上沉積的(h)FIB-SEM圖像,(i)相應的放大SEM圖像,以及(j)側視圖以及(k)頂視圖;
采用X射線光電子能譜(XPS)和氬離子刻蝕技術研究了不同電解質下鎂電極界面層的化學組成,組裝了在0.5mAcm-2下10次循環后循環的鎂-銅電池。首先,作者分析了高熵電解質形成的界面層的成分。N和S是陰離子的典型元素,可以反映TFSI或OTf?在DME和TMP中的還原程度。在Ar離子蝕刻前后,N1s信號可忽略不計(圖3a)。考慮到TFSI是N元素的唯一來源,因此添加LiOTf和TMP限制了TFSI的降解。因此,-SOx(x≤2)的副產物必須來源于OTf?(圖3b)。F1s光譜中685.4eV處的峰(圖3c)、P2p光譜中134.3eV處的峰值(圖3d)與Mg2p光譜中的重疊峰(50.84eV)一致,證明Mg3(PO4)2、MgF2和MgO在Mg表面上共存。近7%的可用P參與界面層,并且大多數P以Mg3(PO4)2的形式存在,這表明TMP在Mg電鍍過程中被廣泛還原,從而對界面層有貢獻。總之,在高熵電解質中形成的外界面層由SOx和MgF2(源自OTf?)、磷酸鹽(源自TMP)、MgO和有機醇鹽組成。
在2分鐘的Ar離子蝕刻后,Mg沉積物上的大量表面離子被去除。即使在2分鐘的Ar離子蝕刻以去除表面離子之后,金屬Mg仍然是Mg2p光譜中的次要離子。這一結果證實了在沒有添加劑的情況下形成的界面層比在有添加劑的條件下形成的層厚得多。此外,TFSI陰離子在基礎電解質中被嚴重還原,正如在N1s光譜所示(圖3e)以及在S2p光譜所示(圖3f)。即使在Ar離子蝕刻之后,這些峰的強度仍然保持,表明TFSI-的存在。與在高熵電解質中形成的界面層相比,在不含添加劑的基礎電解質中形成的界面層更厚,并且含有大量TFSI衍生的物質(MgS、Mg3N2、N-SOx和SOx)和有機物(被DME還原),這會顯著阻斷Mg2+的擴散
元素N、S和F只能來自OTf?或TFSI陰離子;因此,作者總結了它們在所形成的界面層中的總比例,以評估陰離子的還原程度。在含有LiOTf和TMP的電解質中,Mg沉積物表面的N、S和F的總比例為7.25%,隨后在Ar離子蝕刻后降至2.64%(圖3h);基礎電解質界面層中N、S和F的總比例為28.9%,幾乎是高熵電解質中的四倍,Ar離子蝕刻后,該比例仍高達25.6%,表明TFSI在基礎電解質中顯著減少。TFSI的顯著減少與Mg鍍覆過程中的電化學條件高度相關。高電壓滯后(~?2 Vvs.Mg2+/Mg)導致非常強的負電場,這將促進基礎電解質中離解的TFSI的降解。相反,添加TMP和LiOTf后形成的優化界面層顯著降低了電壓滯后,緩解了TFSI-的降低。
利用TOF-SIMS來表征在高熵電解質中形成的界面層。PO4和Mg-P-O的存在有力地證明了Mg3(PO)4是在界面層中形成的(圖3i,j)。利用傅立葉變換紅外光譜(FT-IR)研究了Mg表面界面層的表面化學。如圖3k所示,結果表明添加LiOTf和TMP前后,表面層的光譜顯示出顯著差異。TFSI和DME的信號明顯減弱,Mg3(PO)4(P-O)出現強信號。這一發現證實了添加LiOTf和TMP前后界面層的顯著差異。得益于添加LiOTf和TMP后形成的薄但致密的界面層,富含Mg3(PO)4的層表現出具有快速界面動力學的Mg2+導電性。相反,由于TFSI和DME的顯著降解,基礎電解質中形成的界面層非常厚,含有大量的MgF2、MgS、Mg3N2、MgO和有機物;這些物質顯著阻斷了Mg2+的擴散,并導致緩慢的電化學動力學。
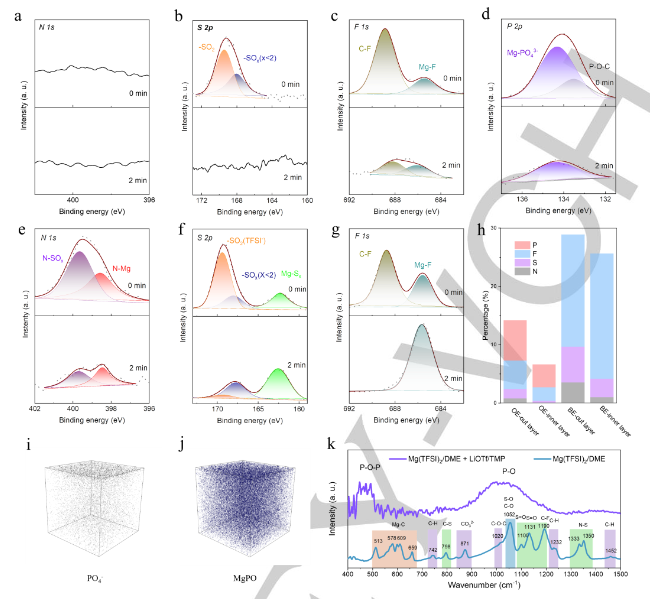
【圖3】Mg(TFSI)2/DME與TMP和LiOTf(優化電解質,OE)形成的界面層的(a)N 1s、(b)S 2p、(c)F 1s和(d)P 2p XPS圖譜。純Mg(TFSI)2/DME(基礎電解質,BE)中形成的界面層的(e)N1s、(f)S 2p和(g)F 1s XPS圖譜。(h)Ar離子蝕刻前后兩種電解質中界面層的原子比。TOF-SIMS深度剖面圖和3D渲染圖揭示了(i)PO4和(j)MgPO在Mg(TFSI)2/DME+TMP/LiOTf中沉積的Mg表面上的分布。(k)在含有和不含有LiOTf/TMP的Mg(TFSI)2/DME中形成的界面層的FT-IR光譜
溶劑化結構
界面層與電解質的溶劑化結構密切相關。因此,作者確定了兩種電解質成分的供體數量,以預測它們的配位狀態(圖4a)。與TFSI-相比,DME分子更容易與純Mg(TFSI)2/DME中的Mg2+配位。相反,OTf和TMP的供體數量相對較高,并且接近DME的供體數量。因此,這兩種添加劑可以參與溶劑化鞘。核磁共振(NMR)結果表明,Li+-TMP和Mg2+-TMP的配位狀態得到證實(圖4b)。對這兩種電解質進行了拉曼表征,可以觀察到了配位TMP分子的存在。向電解質中添加LiOTf后,S=O(1286.8cm-1)的振動模式增加,證明了配位OTf的存在(圖4c)。在Mg(TFSI)2/DME的拉曼光譜中,~881cm-1處的峰值對應于Mg(DME)32+,這與預測一致(圖4d)。在添加TMP和LiOTf后,對應于Mg(DME)32+的峰的消失清楚地表明,由于添加劑的強配位能力,溶劑化鞘層發生了變化。
對Mg(TFSI)2/DME在LiOTf和TMP存在和不存在下的溶劑化結構進行了理論計算。純Mg(TFSI)2/DME中的溶劑化鞘為Mg(DME)32+(圖4e),這與拉曼光譜一致。值得注意的是,靜電勢的正電荷富集均勻分布在DME分子中的C-O鍵上(圖4f),這意味著電子從DME中的O顯著轉移到Mg2+。這種富集將促進DME的還原并形成無活性的Mg(OCH3)2(DME)2物種。因此,Mg表面被大的過電位鈍化,這進一步導致TFSI降解。然而,添加LiOTf和TMP后,溶劑化結構發生了顯著變化。其中一個溶劑化的DME分子被OTf-取代,OTf-橋接Li+形成Mg2+-2DME-OTf--Li+-DME-TMP溶劑化結構(圖4g)。因此,靜電電勢被重新分配。添加LiOTf和TMP后,DME的正電荷富集也發生了顯著變化(圖4h),最終削弱了從O到Mg2+的電子轉移,緩解了DME的還原。此外,這種高熵溶劑化結構將OTf?和TMP帶到了Mg電極的表面,并促進了它們的還原,這有利于構建Mg2+導電界面層。
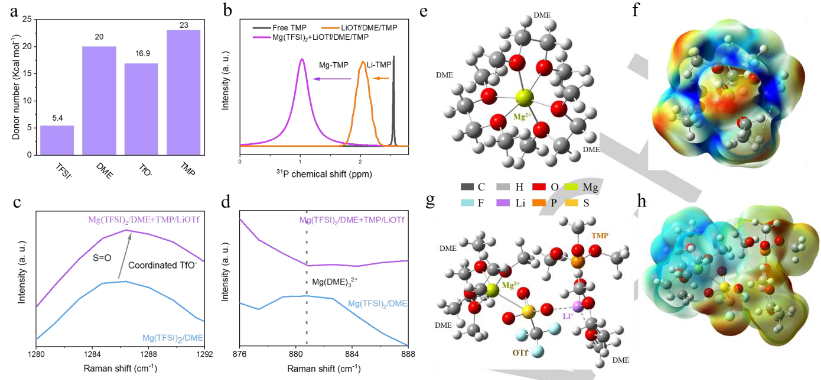
【圖4】(a)TFSI-、DME、OTf-和TMP的供體數量。(b)純TMP溶劑和TMP在不同環境中的31PNMR光譜。(c,d)兩種電解質在(c)1280–1292 cm-1和(d)876–888 cm-1波數范圍內的拉曼光譜。(e)典型的溶劑化鞘和(f)Mg(TFSI)2/DME中相應的靜電電勢分布。(g)典型的溶劑化鞘和(h)具有LiOTf/TMP的Mg(TFSI)2/DME中相應的靜電電勢分布。靜電電勢分布圖中的紅色和藍色區域分別代表負電荷富集區和正電荷富集區
03 Mg||Mo6S8電池的電化學性能
將Mo6S8與鎂金屬負極組裝,以評估高熵電解質的實用價值。如圖5a所示,含有Mg(TFSI)2/DME的Mg||Mo6S8全電池在最初的幾個循環后提供了約2mAhg-1的低放電容量。得益于高熵電解質,Mg||Mo6S8全電池表現出穩定的放電容量和超過200次循環的長壽命。含有Mg(TFSI)2/DME的Mg||Mo6S8全電池的庫侖效率明顯波動(圖5b),而添加LiOTf和TMP的全電池的庫倫效率保持相對穩定,并在200次循環中逐漸達到98.4%。這種效率比對照組的效率高得多。此外,電池的電壓-容量曲線證實,充電/放電過程在高熵電解質中是穩定的(圖S23a)。相比之下,在含有Mg(TFSI)2/DME的Mg||Mo6S8全電池中觀察到約2mAhg-1的高過電位和低容量,這歸因于所形成的絕緣界面層。盡管使用這種高熵電解質的Mg||Mo6S8全電池相對穩定,但這種全電池的衰變趨勢歸因于電解質的消耗和由此產生的電極極化。
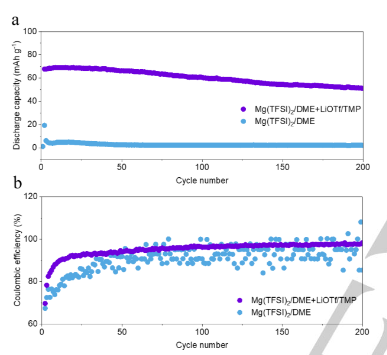
【圖5】(a)Mg‖Mo6S8電池在兩種電解質中的電化學性能和(b)庫侖效率
總結與展望
通過在Mg(TFSI)2/DME電解質中共同添加LiOTf和TMP來穩定Mg金屬負極。具有高供體數的OTf和TMP協同形成了高熵Mg2+-2DME-OTf-Li+-DME-TMP溶劑化結構,從而優化了去溶劑化過程,并在負極表面形成了富含Mg3(PO4)2的Mg2+導電膜。得益于高熵電解質,鎂-銅電池提供了高達98%的高庫侖效率。鎂對稱電池在初始循環期間提供了約80mV的低電壓滯后,在450小時內逐漸增加到580mV。鎂合金Mo6S8全電池的穩定電化學性能證明了高熵電解質設計在無氯鎂金屬電池中的潛在價值。
審核編輯:劉清
-
SEM
+關注
關注
0文章
254瀏覽量
14717 -
電解質
+關注
關注
6文章
821瀏覽量
20496 -
電解液
+關注
關注
10文章
860瀏覽量
23372 -
EIS
+關注
關注
0文章
27瀏覽量
8973 -
TMP
+關注
關注
0文章
15瀏覽量
31928
原文標題:北大徐東升等Angew:高熵電解液賦能高可逆長循環鎂金屬電池
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
鋰離子電池電解液超全面介紹 有何神秘之處?
電解液——鋰電池的‘血液’
新宙邦高鎳三元/硅碳電池電解液批量應用,主要占據中高端市場
解析高電壓水系電解液最新研究進展






 工商網監
工商網監

評論