 詳細描述LAMMPS在Device Studio中的應用
詳細描述LAMMPS在Device Studio中的應用
8.2.LAMMPS實例
LAMMPS即Large-scale Atomic/Molecular Massively Parallel Simulator,大規模原子分子并行模擬器,主要用于分子動力學相關的一些計算和模擬工作。一般來講,分子動力學所涉及到的領域,LAMMPS代碼也都涉及到了。LAMMPS由美國Sandia國家實驗室開發,以GPL license發布,即開放源代碼且可以免費獲取使用,這意味著使用者可以根據自己的需要自行修改源代碼。LAMMPS 程序在模擬固態材料(金屬、半導體)、柔性物質(生物分子、聚合物)、粗粒度介觀體系等方向具有廣泛的應用。LAMMPS 程序內置多種原子間勢(力場模型),可以實現原子、聚合物、生物分子、固態材料(金屬、陶瓷、氧化物)、粗粒度體系的建模和模擬。該程序即可以模擬二維體系,也可以模擬三維體系,可以模擬多達數百萬甚至數十億粒子的分子體系,并提供支持多種勢函數,具有良好的并行擴展性、模擬效率高、計算時間短等優點。
分子動力學模擬(Molecular Dynamics,MD)是近年來飛速發展的一種分子模擬方法,已經被廣泛應用于化學化工、材料科學與工程、物理、生物醫藥等科學和技術領域,起到越來越重要的作用。MD模擬用來研究不能用解析方法來解決的復合體系的平衡性質和力學性質,用來搭建理論和實驗的橋梁,在數學、生物、化學、物理學、材料科學和計算機科學交叉學科占據重要地位。
鴻之微科技(上海)股份有限公司在Device Studio 2021B中開發了適用于分子動力學計算軟件 LAMMPS 的計算模塊。使用Device Studio,用戶可在其圖形界面中方便快捷的搭建或導入計算所需的結構,并可在3D顯示區域查看其結構的3D視圖。搭建好結構后,用戶可在LAMMPS計算模塊,根據計算需要,在簡潔友好的界面中設置參數生成計算所需的輸入文件,之后連接裝有LAMMPS的本地電腦或遠程服務器進行相關計算,在計算過程中可實時監測任務的計算狀態,計算完成后可對LAMMPS的計算結果進行可視化分析。
目前用戶可通過Device Studio生成LAMMPS以下計算輸入文件的生成:結構弛豫、熱力學性質(熱膨脹系數、體積熱容、等壓熱容)、輸運性質(均方位移、速度自相關函數、熱導率)、力學性質(楊氏模量、剪切模量)、淬火、退火模擬;結合OVITO軟件可對LAMMPS計算結果進行結構特征數據分析。
以對金屬鋁沿X軸方向以一定的恒定應變速率拉伸的形變模擬為例來詳細描述 LAMMPS 在Device Studio中的應用。
8.2.1.LAMMPS計算流程
LAMMPS分子動力學計算在Device Studio中的流程如圖8.2-1所示。
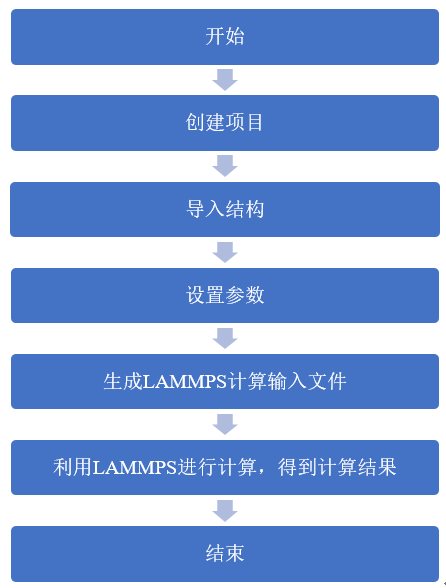
圖8.2-1: LAMMPS計算流程
8.2.2.LAMMPS創建項目
雙擊DeviceStudio圖標快捷方式啟動軟件,根據界面提示選擇創建一個新的項目(Create a new Project)或打開一個已經存在的項目(Open an existing Project)的按鈕,選中之后點擊界面中的OK按鈕即可。若選擇創建一個新的項目,用戶可根據需要給該項目命名,如本項目命名為LAMMPS,或采用軟件默認項目名。
8.2.3.LAMMPS導入結構
在Device Studio的圖形界面中點擊File→Import→Import Local, 則彈出導入LAMMPS結構文件的界面,根據界面提示找到Al_Rede.hzw結構文件的位置,選中Al_Rede.hzw結構 文件,點擊打開按鈕則導入Al_Rede.hzw結構后的Device Studio界面如圖8.2-2所示。
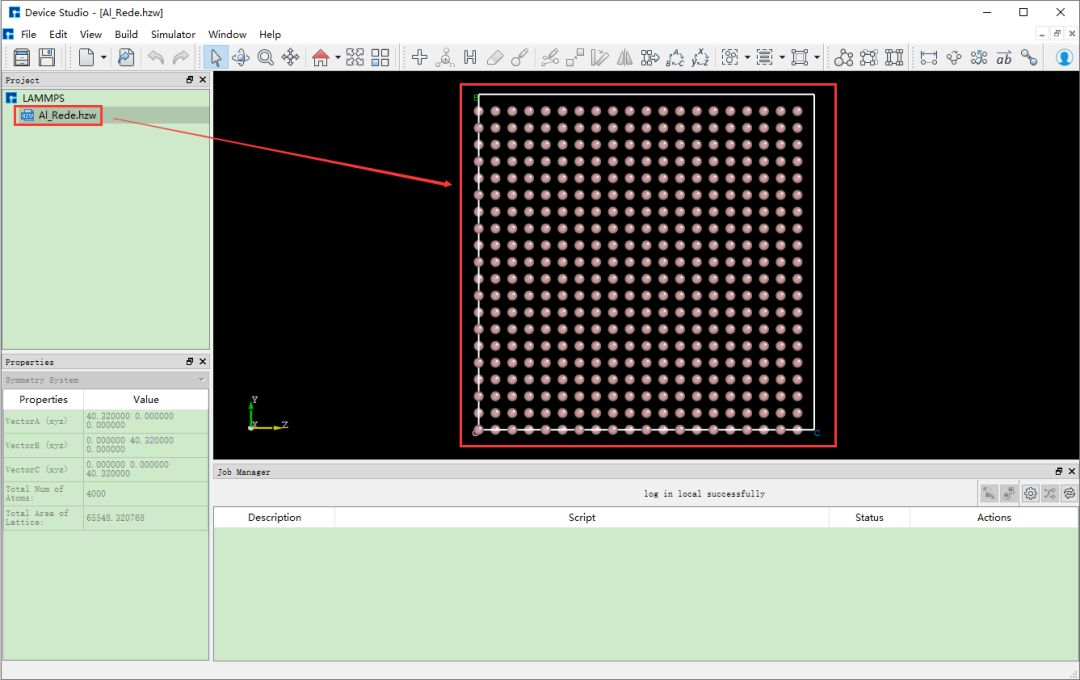
圖8.2-2: 導入Al_Rede.hzw結構后的Device Studio圖形界面
審核編輯:劉清
-
計算機
+關注
關注
19文章
7511瀏覽量
88134 -
仿真器
+關注
關注
14文章
1018瀏覽量
83790 -
模擬器
+關注
關注
2文章
877瀏覽量
43257
原文標題:產品教程丨多尺度材料設計與仿真平臺Device Studio(應用實例05)
文章出處:【微信號:hzwtech,微信公眾號:鴻之微】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
哪里有ps7-gpio的設備樹文檔的不同屬性的詳細描述?
RTC實時時鐘詳細描述
DS1302芯片詳細描述
詳細描述RTT QSPI驅動配置過程
有誰知道哪里有Sequencer的詳細描述以及示例?
多層印刷電路板及產品詳細描述
常用三種指針的設計類型詳細描述
KeyStone中使用PCIE的應用案例和PCIE特征的詳細描述






 工商網監
工商網監
評論