 鋰離子電池表面改性正極的快速嵌鋰機理
鋰離子電池表面改性正極的快速嵌鋰機理
研究背景
利用氧化材料(如Li2ZrO3)對正極表面進行改性,鋰電池正極的循環穩定性和倍率性能得到顯著提高。部分研究指出,改性層的成分通過與電解質發生電化學反應而改變,促進離子導電CEI層的形成,為進一步了解電池運行下界面結構的變化,以及改性正極-有機電解質界面上鋰(脫)嵌入的反應速率,必須使用單一的技術來研究電池反應過程中從電極側到電解質側的界面結構變化。
成果簡介
日本東京工業大學Masaaki Hirayama教授利用原位NR研究Li2ZrO3改性LiCoO2正極和未改性正極,發現表面改性有利于形成致密的、由無機物種組成的CEI,未改性的表面覆蓋著相對稀疏且浸漬了電解液的CEI。研究發現,鋰在插層過程中的脫溶主要發生在CEI和LiCoO2表面,鋰在CEI上的快速溶解可能有助于表面改性的正極具有優異的倍率性能,提出了一種通過利用低離子導電性氧化物進行表面改性來實現快速插層的機制。該工作以“Fast Lithium Intercalation Mechanism on Surface-Modified Cathodes for Lithium-Ion Batteries”為題發表在Advance Energy Materials上。
研究亮點
(1)用PLD方法在SrRuO3(100)/SrTiO3(100)襯底上外延生長了LiCoO2(104)和Li2ZrO3改性的LiCoO2(104)薄膜。
(2)與未改性的LiCoO2(104)相比,改性的LiCoO2具有更好的鋰插層速率能力。
(3)改性表面的內層由無機物質組成,未改性的表面被相對稀疏的電解質浸漬的CEI覆蓋,與LiCoO2表面相比,改性LiCoO2優越的速率能力可能源于CEI上鋰能更快脫溶,并穩定電極表面,為適合大功率工作的正極-液-電解質界面設計提供了一種新的設計原則。
(4)利用原位NR直接觀察循環過程中的界面結構,能夠同時檢測化學成分和形態信息,是闡明電池反應詳細機理的有力方法。
圖文導讀
圖1為在SrRuO3(100)/Nb:SrTiO3(100)上合成的未改性和Li2ZrO3改性的LiCoO2(104)薄膜的XRD圖和XRR譜圖。層狀巖鹽型LiCoO2沿SrTiO3<100>、<010>和<011>方向分別有104、01-4和01-8衍射峰。這證實了未改性和Li2ZrO3改性的LiCoO2薄膜的外延生長。Li2ZrO3沉積過程中沒有形成固溶體,這歸因于Zr4+在層狀巖鹽型結構中的溶解度差。未改性和改性的LiCoO2膜的厚度分別為22.1 nm和22.0 nm, SLD值分別為38.1×10?4和38.3×10?4 nm,與XRD結果一致,表明未形成LiCoO2-Li2ZrO3固溶體,兩種樣品的表面粗糙度均為≈1 nm,表面面積無顯著差異。因此,未改性和Li2ZrO3改性的LiCoO2(104)薄膜適合研究改性對正極-電解質界面電化學反應的影響。
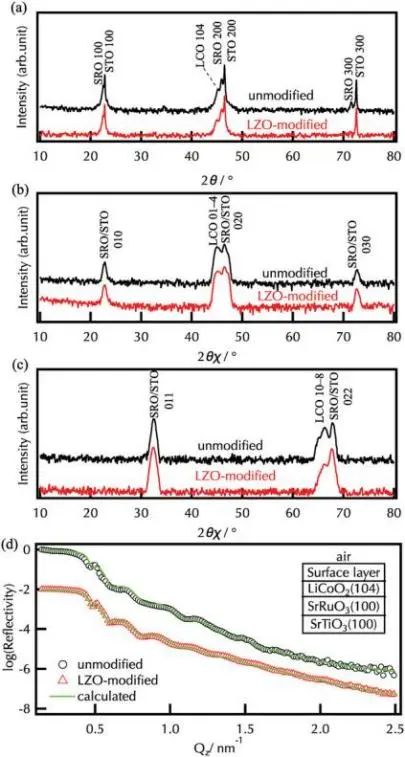
圖1. a) SrRuO3(100)/Nb:SrTiO3(100)上未改性和Li2ZrO3改性的LiCoO2膜的a)面外、b,c) 面內XRD譜圖和d) XRR譜圖。LCO、LZO、SRO、STO分別代表LiCoO2、Li2ZrO3、SrRuO3、SrTiO3。擬合模型和計算得到的XRR譜如圖1d所示。
在100C(300μA cm?2)條件下,兩種膜的放電容量相近,約為125 mAh g?1,相當于LiCoO2的可逆鋰插層。高容量保持是因為電子和鋰離子在26 nm厚的薄膜電極內的傳輸距離較短。未改性薄膜的Vave在第一次循環時放電電壓為3.53 V,到第五次放電時Vave值逐漸下降到3.49 V,這是由于放電結束時電壓降低,在之后循環中 Vave 值隨著電流密度降低而增加。相反,改性薄膜的放電電壓較高,達到3.80 V,在100C下的五個循環中沒有明顯下降。XRD分析證實,未改性和改性薄膜的晶體結構在生長狀態下沒有發生實質性變化,此外,在20次充放電循環過程中,高度可逆的插層容量表明26 nm厚的鈷酸鋰表面沒有發生實質性退化。因此,與未改性的薄膜/電解質界面相比,Li2ZrO3改性的LiCoO2/電解液界面表現出更高的鋰插層反應速率。
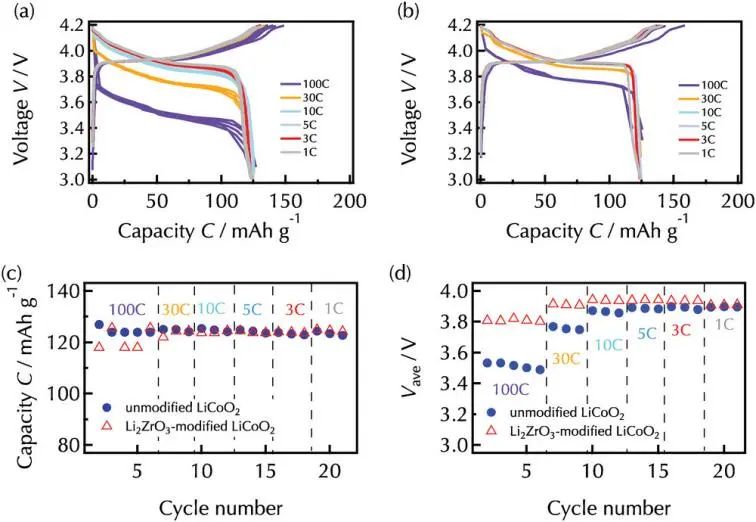
圖2. a)未改性LiCoO2和b)Li2ZrO3改性LiCoO2在100C(300μA cm?2)到1 C(3μA cm?2)不同的放電電流密度下充放電曲線,充電電流密度固定為1C。c)放電容量的變化和 d)從(a、b)中獲得的平均放電電壓。在100C下改性LiCoO2放電容量的變化是由于充放電設備的時間分辨率所致。
圖3是兩種薄膜在第二個周期中不同放電電壓下的EIS圖。對于這兩種薄膜,在4.1V、4.0V和3.9V都有明顯的扭曲半圓,其直徑從4.0增加到3.9V。在3.7V和3.5V時,10 Hz以下的低頻區阻抗值急劇增加,而在10Hz以上觀察到的半圓比例隨著電壓的變化而相對保持不變。這表明,3.9V以上的扭曲半圓由兩個時間常數相近的分量組成。因此,作者使用具有RLFCPELF和RHFCPEHF兩個RC分量的等效電路來改進Nyquist曲線圖。
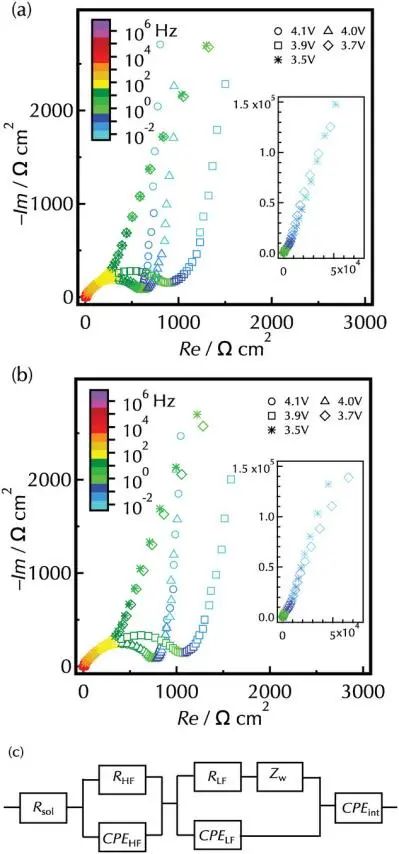
圖3. a)未改性的LiCoO2和 b)Li2ZrO3改性的LiCoO2薄膜在第二個循環中放電至不同電壓后的奈奎斯特圖。c) 采用等效電路進行頻譜擬合。
在第二次充放電循環期間,未改性膜和改性膜的RLF值在充電時從3.9V下降到4.1V,放電時從4.1V可逆地增加到3.9V。由于(脫)嵌鋰速率會隨著正極材料中Li+濃度的變化而變化,因此RLF與正極-電解質界面上鋰(脫)插鋰的界面電阻有關。與RLF相比,兩種薄膜的RHF值隨電池電壓的變化而保持相對穩定,考慮到電解液和鋰正極中包括的電化學過程中的微小電阻,正極側可以包括另一個具有高時間常數的電阻分量,如果假定Li2ZrO3基CEI層的離子電導率與Li2ZrO3相當,則計算出CEI層的鋰轉移電阻為≈15Ω cm2,這導致在100C工作時極化為5 mV,RHF不能確定為RCEI。因此,在高頻下,電子接觸電阻Re可能是半圓的主要電阻成分。總體而言,兩種薄膜在所有條件下的 RLF 值都高于RHF值,這意味著在薄膜電池中,正極界面的鋰插層是決定速率的步驟。正極-CEI-電解質界面包含了鋰嵌入的幾個基本過程:i)溶劑化鋰在CEI表面的吸附和脫溶,ii)鋰在CEI層中的轉移,以及iii)鋰嵌入伴隨著正極-CEI界面的電子轉移。過程(i)和(ii)的速率可能取決于CEI層的化學成分和結構,這些成分和結構會因表面改性而改變。鋰嵌入過程的速率(iii)取決于反應速率常數和正極和電解質兩側界面處的鋰濃度,作者假設未改性和改性的LiCoO2薄膜表面沒有明顯的劣化,因為在4.2V和3.0V電壓區域循環過程中充放電容量保持一致。這表明鋰嵌入過程的速率主要取決于電解質側界面。
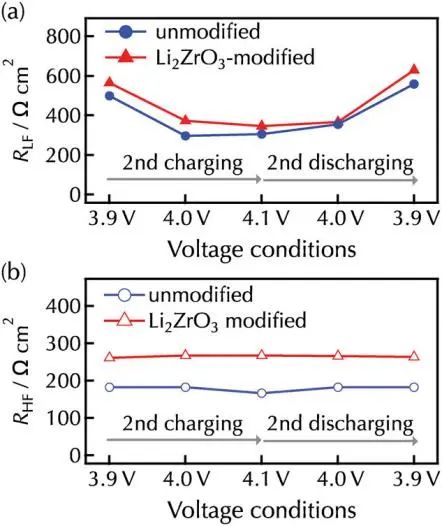
圖4. 未改性和li2ZrO3改性的LiCoO2在第二次充放電時的a) RLF和b) RHF值的變化
圖5為未改性和Li2ZrO3改性的LiCoO2(104)薄膜在第三次放電后的NR譜擬合結果和nSLD曲線,與原始薄膜相比,由layer1/layer2/LiCoO2/SrRuO3/SrTiO3組成的雙表面層模型與兩種薄膜的反射率曲線非常吻合。未改性和改性的LiCoO2層均表現出相似的nSLD和粗糙度,這表明未改性和改性的LiCoO2之間沒有明顯的結構差異。對于未改性的LiCoO2,外層和內層的厚度分別為2.2nm 和1.5nm,改性后的LiCoO2包覆了厚度為1.5nm的外層和內層,nSLD分別為3.09×10?4nm?2和2.42×10?4 nm?2,據報道,原始狀態下的表面層被主要由電解質物質分解形成的CEI物質所取代,考慮到CEI成分的nSLD 值,外表層主要由Li2ZrO3和 CH2OCO2Li組成,底表層主要由 LiF、Li3POxFy和 LiOH組成。改性表面內層的nSLD值略高于未改性表面,可能是由于基于Li2ZrO3的成分,如 Li2ZrOxFy。雙表面層模型與先前的研究一致,表明無機CEI物質最初在正極表面形成,然后在正極表面堆積有機CEI物質。NR和XPS分析均檢測到未改性和改性LiCoO2表面之間CEI層的成分或厚度沒有顯著變化。
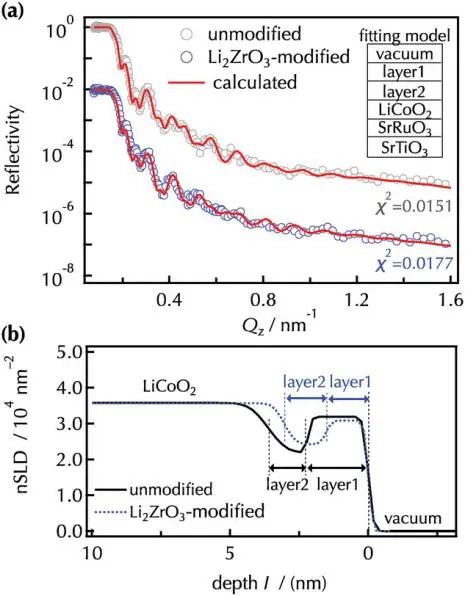
圖5. a)觀察并計算第三次放電后未改性LiCoO2和Li2ZrO3改性LiCoO2的NR光譜;b)細化了未改性和Li2ZrO3改性LiCoO2薄膜的nSLD曲線。采用SrRuO3/LiCoO2/layer2/layer1雙表面層模型計算非原位NR光譜.
為消除采樣過程對實驗結果造成否影響,作者進行了原位NR分析,以探索初始反應過程中的電極-電解質界面。圖6和圖7顯示了電池構建后以及在第三個循環中充電和放電狀態下未改性和Li2ZrO3改性的LiCoO2(104)薄膜的原位NR擬合結果和nSLD曲線。與非原位NR分析一致,雙界面層模型(SrRuO3/LiCoO2/內層/外層/電解液)與兩個樣品的反射率光譜密切相關。4.2V的帶電狀態下,未改性和改性的LiCoO2層的nSLD值高于電池構建后的觀察結果,這對應于缺鋰相Li1?xCoO2的形成,這可能是由于充電過程中鋰的脫嵌作用。在放電狀態下,nSLD值可逆地減小,證實了鋰嵌入到Li1?xCoO2中,在兩種薄膜的充放電循環過程中,LiCoO2層(≈分別為25 nm和1 nm)的厚度和粗糙度保持穩定,只有很小的變化。這些發現表明,可逆的鋰(脫)插層發生在不降解LiCoO2相的情況下,與在充放電曲線中觀察到的高容量保留一致。
電池構建后,在未改性的LiCoO2表面形成了厚度為2.6nm的內層,在充放電過程中,沒有觀察到明顯的厚度、nSLD 或粗糙度變化,這表明,浸入電解液后形成的內層在隨后的過程中保持電化學穩定。以前的研究表明,LiOH和Li2ZrO3等表面物種與電解液中的HF和H3PO4等雜質發生化學作用,生成LiF和Li3PO4,從而形成無機CEI層。然而,第三次放電后觀察到的nSLD值遠遠高于理論值,電解液的nSLD值為5.67×10?4 nm?2時,可能會使nSLD值增大,因為電解液可能會填滿CEI層內的空洞。因此,在未經改性的表面上形成的內層可能既包括無機CEI物種,也包括液體電解液。在Li2ZrO3改性的LiCoO2表面,電池的內層厚度在第三次放電后變化很小,這表明內部的CEI是在浸泡在電解液中后形成的,這是由于Li2ZrO3與電解液物質之間發生了化學反應。nSLD值顯著低于未改性LiCoO2,且與非原位NR測得的改性LiCoO2的內部CEI值吻合,表明改性的Li2ZrO3層促進了致密的無機CEI的形成,孔洞相對較少,在電化學條件下,這種致密的CEI有助于液體電解液向內層的滲透率。因此,原位NR分析表明,表面改性既影響CEI層的密度,也影響液體電解液的浸潤量,這些可能會改變鋰離子在插層過程中的界面擴散過程。
在未改性和Li2ZrO3改性的LiCoO2中觀察到明顯的外層,厚度超過5nm,在電化學條件下,外層表現出不同于在干燥條件下觀察到的不同成分和形態。未改性的LiCoO2表面外層在充放電過程化學成分發生了變化,相反,改性LiCoO2的外層顯示出更低的循環前nSLD值,為3.46×10?4 nm?2,在4.2V時,nSLD值增加到3.69×10?4 nm?2,而在3.2V時,nSLD值基本不變,與內層類似,改性前后外層界面結構的變化不同。作者提出了三種可能的界面模型來解釋外層傾斜的nSLD分布:i)具有成分梯度的CEI層,ii)電解液中離子濃度的分布,以及iii)固體CEI與液體電解質比率的深度變化。在模型(III)的基礎上,作者探討了充電和放電過程中外層的結構變化。對于未改性的LiCoO2,當充電到4.2V時,nSLD增加,表明在外層形成了有機CEI,放電3.2V時nSLD的減少表明有機物種的分解或去除。Li2ZrO3改性的LiCoO2表現出比未改性的變體更低的nSLD,這表明在外層有更高的無機/有機CEI比和液體電解液,值得注意的是,外層在充放電過程中nSLD和厚度沒有顯著變化,模擬了內層的行為。在充放電的初始階段,改性LiCoO2的界面結構建立了一定的化學成分和形態。致密的內部CEI阻礙了LiCoO2與液體電解液之間的電子接觸,從而阻礙了電解液物種的進一步分解,從而擴大了CEI層。相反,未改性的LiCoO2形成的內部CEI密度較低,導致循環過程中外部CEI的生長和分解。
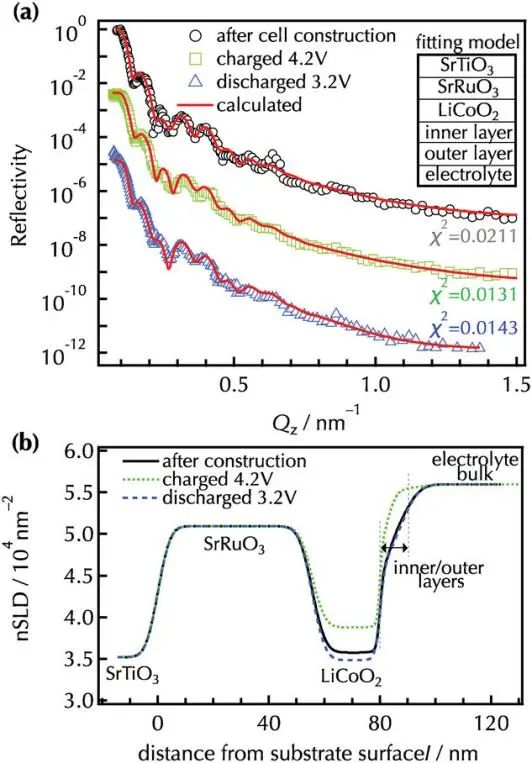
圖6. a)觀察和模擬了未改性LiCoO2薄膜的NR譜,b)改進了電池構建(3.3V)和充電到4.2V后第三次循環放電到3.2V后的nSLD曲線。用SrRuO3/LiCoO2/內/外層四層模型計算了原位NR光譜.
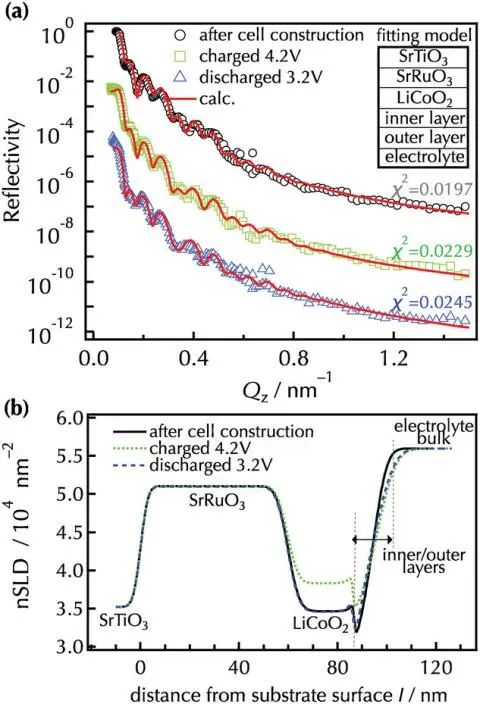
圖7. a)觀測和模擬了Li2ZrO3改性的LiCoO2薄膜的NR譜和b)精化的nSLD譜。用SrRuO3/LiCoO2/內/外層四層模型計算原位NR光譜.
對于這兩種類型的LiCoO2,根據從LiCoO2表面到電解液本體的距離,界面結構分為內部和外部。內層和外層分別由無機CEI和有機CEI和電解液的混合物組成。Li2ZrO3的表面改性有利于無機CEI的形成,導致內層有致密的無機CEI,外層有較高的無機/有機CEI比值。基于這些關于界面結構的發現,作者預測鋰插層過程中的鋰轉移過程如下:
未改性的LiCoO2:內層和外層的空腔浸漬有液態電解質。鋰離子可在電解液中擴散,電解液的鋰傳導性大大高于CEI元件。鋰離子從電解液快速穿過界面層,到達鈷酸鋰。然后離子脫溶,被鈷酸鋰表面吸收,接著插層進入鈷酸鋰晶格中。
Li2ZrO3改性LiCoO2:鋰離子通過液體電解液擴散,浸漬在外層,到達內表面。由于內部CEI的高密度,大多數鋰離子必須去溶才能擴散到內層。脫溶的鋰離子通過LiCoO2和CEI內部的固-固界面嵌入到LiCoO2晶格中。
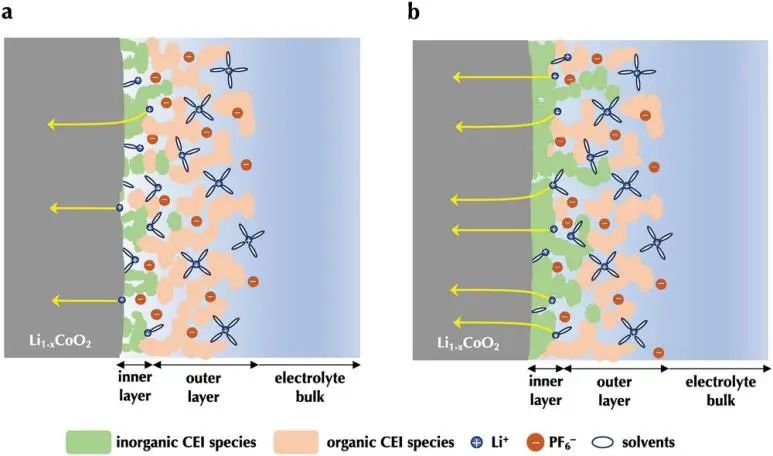
圖8. a)未改性和b)Li2ZrO3改性的LiCoO2在放電條件下的電極/電解液界面示意圖。
總結與展望
在改性表面的內層是由無機物種組成的,這有利于鋰在內層CEI上的插層脫溶。相反,未改性的表面覆蓋著相對稀疏且浸漬了電解液的CEI,脫溶主要在LiCoO2表面進行。因此,與LiCoO2表面相比,改性LiCoO2優異倍率能力可能源于CEI上鋰的脫溶速度更快,而這種變化是無法在原位跟蹤的。然而,這些變化應該與循環開始時的界面結構有關。作者強調了表面改性在促進鋰離子快速解溶的界面結構方面所起的以前未明確的作用,從而導致更高的鋰插層速率。這一發現將為表面改性材料的設計提供新的概念。
-
鋰離子電池
+關注
關注
85文章
3254瀏覽量
77865 -
正極
+關注
關注
0文章
53瀏覽量
10335 -
電解質
+關注
關注
6文章
820瀏覽量
20123
原文標題:日本東京工業大學Masaaki Hirayama教授:鋰離子電池表面改性正極的快速嵌鋰機理
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦





 工商網監
工商網監
評論