 優化碳酸酯基電解液用于鋰負極
優化碳酸酯基電解液用于鋰負極
研究背景
鋰離子電池的改進對于實現車隊的廣泛電動化至關重要。目前,實現這一目標的最大障礙之一是缺乏價格合理的高能量密度電池。用鋰金屬負極解決了這些問題,使其成為該領域日益受到研究的話題。適當選擇電解質對于確定電池的安全性、容量和循環壽命至關重要,尤其是在使用鋰金屬負極時。液體電解質的最大優點之一是組分(鹽、溶劑和添加劑)易于合成,具有所需的功能化以改變 SEI 結構和組成。不幸的是,用碳酸酯電解液循環的鋰金屬負極上產生的SEI是厚的,多孔的,不均勻的,對體積變化很脆弱,并且無法抑制枝晶生長,導致與使用醚基電解質相比觀察到的性能較差。因此,通過選擇改性碳酸酯基電解質,可以在很大程度上保持對高壓正極的氧化穩定性,同時可以形成由所需產物組成的SEI,以產生能夠在各種電位下運行的能量密集型鋰電池。
成果簡介
近期,羅德島大學Brett L. Lucht教授團隊通過創新性地對碳酸酯基電解質進行的不同優化,包括不同類型和濃度的鹽和添加劑,以產生具有有益特性并提高鋰電池整體性能的SEI。未來的電解質設計選擇可以針對電池的應用定制碳酸酯基電解液。
該工作以“Modification of Carbonate Electrolytes for Lithium Metal Electrodes”為題發表在期刊ACS Energy Lett上。第一作者為羅德島大學Munaiah Yeddala。
研究亮點
(1)研究不同鋰鹽和溶劑的組合對電極的優化
(2)研究不同溶劑電化學反應路徑與界面行為
(3)提出新型的添加劑并對現有碳酸酯電解質進行展望
圖文導讀
圖1是本次實驗所用的鋰鹽與添加劑的結構與化學式
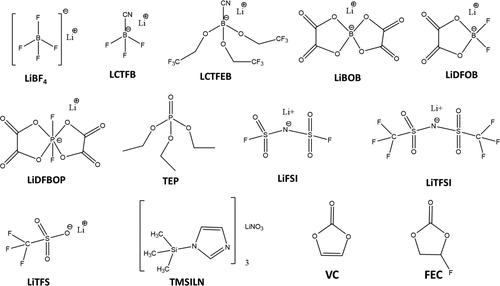
圖1 本觀點中討論的鋰電池電解質中使用的鹽和添加劑的化學結構
環狀碳酸酯
環狀溶劑的一般分解反應如圖2a–c所示。環狀碳酸酯通常包含在電解質混合物中,因為正極表面的穩定性提高,鋰鹽的強溶劑化以及負極表面的良好鈍化。EC和PC很容易在電壓~0.8-1.0 V與Li/Li下發生單電子開環反應,這與SEI形成的初始階段相吻合。雖然EC還原的確切機制仍在爭論中,但在SEI中形成的初始還原產物是二碳酸乙烯鋰(LEDC)和乙烯氣體。雖然由于電極不穩定,通常避免在石墨負極上使用PC,但PC可用于鋰金屬負極以形成類似的還原產物,即二碳酸鋰丙烯(LPDC)和丙烯氣體。
隨著循環的繼續,已知由這些組分形成的SEI會演變和變化,這可能是由于鋰金屬負極上的烷基碳酸鋰LEDC和LPDC的不穩定性。在熱、微量水或一些鋰鹽存在下,LEDC可以分解溶解以增加SEI的孔隙率,從而導致電解質持續還原生成Li2CO,以及其他與鋰鹽相關的還原產物。這導致在循環電池的負極上多層SEI的產生,其內部SEI由無機物質組成,包括LiF。與EC或PC不同,FEC是一種環狀碳酸酯,由于在相對較高的電位1.1 V與Li/Li下還原,因此可用作助溶劑或添加劑。
在含FEC的電解質中,TEM分析顯示鋰負極上形成均勻的納米結構SEI(圖2d),EDX分析顯示SEI由覆蓋LiF納米顆粒組成和一氧化碳,LiF納米顆粒導致均勻的擴散場,導致鋰的更均勻的電鍍和剝離,這也抑制了Li枝晶的生長。隨著循環的繼續,FEC的還原也產生了一系列有機物質,覆蓋任何最初沉積的LiF,形成外部聚合物有機SEI層,該層比碳酸酯電解質混合物通常觀察到的更薄。
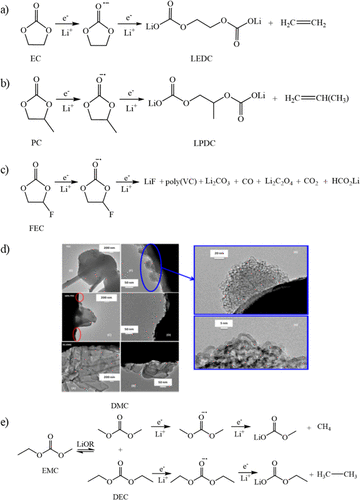
圖2. a)碳酸乙烯酯、(b)碳酸丙烯酯和(c)氟碳酸乙烯酯的廣義分解反應。(d) 在FEC電解質中形成的納米結構SEI的TEM圖像。(e) 碳酸甲乙酯、碳酸二甲酯和碳酸二乙酯的廣義分解反應。
線性碳酸酯
電解質中的線性碳酸酯與環狀碳酸酯具有不同的用途,因為它們具有較低的熔點和較低的粘度,當與EC或PC混合時,在電化學穩定性窗口中具有更高的離子電導率和協同效應。在線性碳酸酯中,最常用的溶劑是DMC,DEC和EMC。DMC和DEC都經歷了類似的還原過程,唯一的區別是碳鏈長度,分別生成CO,甲烷/乙烷,甲醇鋰/乙醇鋰和碳酸甲酯鋰/碳酸乙酯鋰。由于分子的不對稱性,EMC可以與電解質中的鋰醇鹽進行酯交換反應以形成DMC和DEC,從而產生產物的混合物,包括CO,甲烷,乙烷,碳酸甲酸鋰,碳酸乙酯鋰,甲醇鋰和乙醇鋰(圖2e)。雖然溶劑中線性碳酸酯的選擇不會對電池的電化學性能產生很大影響,但研究表明,產生的SEI通常由所用環狀碳酸酯的還原產物而不是線性碳酸酯還原產物主導。因此,任何為改善鋰負極循環性能而定制SEI的電解質修飾都應主要集中在對環狀碳酸酯或鹽的調整上,或者摻入添加劑或其他助溶劑,這些添加劑或其他助溶劑將優先減少以形成更穩定的SEI。
碳酸酯的缺點和替代品
與鍍鋰和剝離相關的體積變化是SEI產品演變為多層結構的主要原因。由標準碳酸酯電解質形成的SEI相當剛性,因此隨著鋰電鍍和剝離的繼續,SEI無法膨脹以適應體積變化,導致SEI出現裂紋。這會將新鮮的鋰暴露于新的電解質中,隨著更多分解產物的形成和演變,導致電解質持續降解和更厚的SEI。據報道,鋰金屬電池中SEI膨脹的證據,導致電解質與鋰表面緊密接觸,鋰腐蝕和電解質耗盡。然而,靠近表面的富含無機物的物種導致更少的腫脹和更好的整體電池性能。
此外,表面上不均勻層的存在會導致鋰的非均勻沉積,最終導致鋰枝晶形成和隨后的電池失效,如果枝晶穿透隔板并使電池短路。由碳酸酯形成的SEI也可以具有絕緣性能,導致形成死鋰,其中未反應的金屬鋰完全包裹在較厚的SEI層中,但與負極中的導電性斷開,因此無法參與電化學剝離或電鍍。枝晶和死鋰的形成如圖3a所示,它們是鋰金屬負極電池失效的重要因素。因此,SEI的柔韌性和穩定性是減少枝晶形成和死鋰以及提高鋰金屬負極容量保持性的關鍵。
除碳酸酯溶劑外,其他最常用的鋰金屬負極電解質是醚基電解質系統。雖然不是本觀點的重點,但使用各種醚(如二甲氧基乙烷(DME)或1,3-二氧戊環(DOL))在提高鋰金屬負極的循環壽命、鋰金屬電鍍和CE性能方面已經取得了重大進展。這主要是由于形成的高度柔韌性的SEI,由LiF和Li的內層組成。和外層含彈性體的聚醚,如圖3b所示。這種聚合的SEI對鋰的體積變化更靈活,并且能夠通過抑制枝晶的形成來延長鋰金屬負極的壽命。然而,醚在高電位下的不穩定性極大地限制了它們在具有高壓正極的高能量密度全電池中的應用。因此,對碳酸酯基電解質中的不同組分進行簡單修改,在SEI中提供與醚基電解質相似的柔韌性。
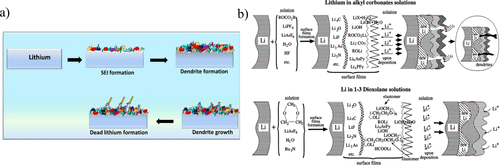
圖3. (a) SEI中枝晶生長和死鋰的示意圖。(b) 碳酸酯和醚基電解質中金屬鋰上的SEI示意圖。
碳酸酯電解質的改性
鹽的選擇
為電解質選擇的鹽是構成SEI的分解產物的重要貢獻者。例如,一些鹽在第一個鍍鋰循環中優先減少,而另一些鹽僅在地層循環完成后才開始分解。因此,選擇與鋰金屬負極循環的鹽是一種重要的改性,可以極大地影響電池性能。
一項對Li/Cu電池的研究了九種不同的鹽(六氟磷酸鋰(LiPF6)、四氟硼酸鋰(LiBF4)、六氟砷酸鋰(LiAsF6)、高氯酸鋰), 雙(三氟甲磺酰)亞胺鋰 (LiTFSI), 三氟甲磺酸鋰、雙(草酸)硼酸鋰 (LiBOB)、二氟(草酸)硼酸鋰 (LiDFOB) 和碘化鋰 (LiI)),并將DOL摻入每種電解質(現在的PC:DME:DOL(1:1:1,v / v)),發現LiPF6的電池電阻增加了,但LiBOB電池(LiBOB-2)的阻抗降低了,如圖4a所示。對這種現象的一種可能的解釋是,將DOL添加到LiBOB溶液中可以防止鹽的繼續分解反應,而LiPF6的路易斯酸度實際上促進了DOL的聚合,從而促進了電解質的消耗。因此,只要以某種方式控制分解程度,LiBOB就能夠形成均勻的SEI層。
基于觀察到的LiBOB作為鹽的改善性能,對鹽中含氟和/或草酸鹽的部分產生穩定SEI的益處進行了進一步的研究。為此,Cu / LFP電池用1.2M LiPF6、1.2 M LiDFOB、0.6 M LiBOB 或 0.6 M LiBOB + 0.6 M LiBF4在相同的EC:EMC(3:7,v/v)溶劑中。有趣的是,1.2 M LiDFOB電解質在所有電解質中具有最佳性能,甚至優于LiBOB / LiBF4電解質,盡管在SEI中具有相似的化學成分。由LiDFOB分解形成的SEI含有納米結構的LiF(5-10nm),這是草酸鹽離子作為封端劑的結果,從而產生均勻的擴散梯度并允許均勻的鍍鋰,如圖4c所示。相比之下,LiBOB / LiBF4電解質產生更大的LiF顆粒(>300 nm)和不均勻,不穩定的SEI,因為LiBOB不斷分解,導致循環不良。這表明SEI的納米結構和形態在鋰剝離/電鍍中起著比通常考慮的更重要的作用。LiDFOB電解質的進一步優化是在Cu / LFP電池上進行的,這些電池已經用含有不同比例LiP6F的EC:DMC或FEC:DMC(1:4,w/w)電解質循環和 LiDFOB 加起來達到 1.0 M。在含有EC:DMC的電池中,性能最佳的電池含有1.0 M LiDFOB,而具有FEC:DMC溶劑混合物的最佳電池含有0.05–0.10 M LiDFOB和0.95–0.90 M LiPF6。圖4b中的紅外光譜表明,SEI中由含LiDFOB的電解質產生的主要物質是Li2C2O4以及納米結構的LiF。低濃度LiDFOB,LiPF6的組合作為主要鹽,FEC作為助溶劑大大提高了鋰金屬負極的電化學性能,從而證實了多種SEI形成組分之間的協同作用。
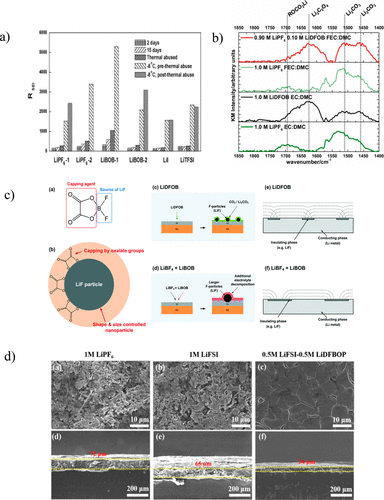
圖4. (a) 不同電解質儲存在不同時期和不同溫度下的Li/Li對稱電池的阻抗。(b) 在含LiDFOB的電解質中循環的鋰電極的紅外光譜。(c) 含 LiDFOB 電解質中的納米結構 SEI 形成機制經參考文獻許可轉載(57).版權所有 2018 英國皇家化學學會。(d) 雙鹽電解質中鋰電極上SEI的SEM圖像(俯視圖和橫截面)。
鹽的混合物
由于多種SEI形成組分已被證明是有益的,特別是在層狀和穩定的SEI的受控生成中,因此利用兩種鹽的混合物可以產生協同效應并提高鋰金屬負極的容量保持率。因此,制備了含有0.5 M LiFSI(以減少電壓極化)和0.5 M二氟雙(草酸)磷酸鋰(LiDFBOP)(優先還原并形成有益SEI產物)的電解質在EC:FEC(3:7,w/w)中,并用Li/Li和Li/LFP電池循環。使用雙鹽電解質循環的Li/Li電池在1000小時內具有出色的長期循環性能,而使用EC:EMC(1:3,w/w)和7 M LiPF6中1 M LiFSI構建的電池在EC:EMC(3:7,w/w)分別在電鍍和剝離僅500和350小時后開始表現出增加的極化。性能的差異可以部分歸因于由每種電解質形成的SEI的微觀結構,如圖4d所示。雙鹽電解質形成致密、光滑、均勻的SEI,有助于防止電解質持續還原并抑制枝晶和死鋰的產生,而單鹽電解質產生多孔、不均勻和厚實的SEI,導致鋰沉積不均勻。因此,如果鋰金屬表面的預處理可用于產生類似的納米結構的層狀SEI,那么在不顯著消耗電解質的情況下,可能會提高性能。然后,通過仔細添加其他協同成分(例如其他鹽或添加劑),可以進一步提高循環性能。
新型添加劑合成
當預先存在的添加劑具有局限性時,例如LiNO3的溶解度差但性質好,可以合成具有相似結構或產生相似分解產物的新型添加劑。例如,合成了一種新的路易斯堿加合物1-三甲基硅基咪唑硝酸鋰(TMSILN)以摻入LiNO3。作為添加劑進入電解質中,無需額外的組分來增加溶解度。由于氮部分與鋰原子的配位,TMSILN可溶于高達1.0wt%。不含添加劑的電解液Li/Li對稱電池在200 h后出現突然的電壓極化,但含有1 wt%TMSILN的電解液在500 mA/cm的電流密度下穩定循環超過1 h。在Li/NCM622電池中應用這種電解質會產生類似的結果,含TMSILN的電解質在90次循環后保留約200%的容量,而基線電解質在第80次循環中僅保留183%的容量(圖5a)。使用TMSILN電解質的第一個循環的差分容量分析表明,與基線電解質相比,添加劑的過電位較低,這意味著電阻較低,這可能是由于LiNO3分解導致電導率提高從加合物。TMSILN添加劑還會影響電池中鋰的物理沉積,其中基線電解質在裸銅箔上產生不均勻的枝晶,但TMSILN電解質沉積球形鋰成核,導致更有利的鋰沉積(圖5a的插圖)。使用TEM和XPS對負極表面的進一步分析表明,含有氮分解物質的SEI生長薄層。因此,從LiNO3獲得的許多有益特性仍然可以使用新型加合物添加劑來實現,同時避免溶解度限制等潛在缺點。
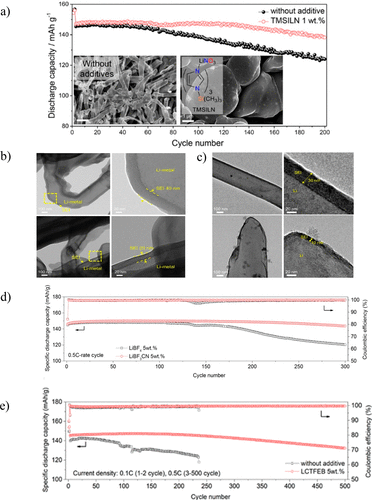
圖5. a) 含有1M LiPF6的Li/NMC532電池的電化學性能在EC:EMC(3:7體積%)電解液中,帶和不帶LTMSILN。插圖顯示了用 1 M LiPF6 循環電解鋰的 SEM 圖像在EC:EMC(3:7體積)電解液中。(b) 鋰電沉積在LiBF中的TEM圖像4-含電解質(頂部)或含LCTFB的電解質(底部)。(c) 鋰電沉積在 1.0 M LiPF 中的 TEM 圖像6在EC:EMC(3:7,v/v)中,不帶(頂部)和5wt%LCTFEB(底部)。(d) 含有 5 wt% LiBF4 的 Li/NMC532 電池的循環數據電解質和 5 wt% LCTFB 電解質。(e) 含有不含添加劑的標準電解質和 532 wt% LCTFEB 的 Li/NMC5 電池的循環數據。
另外,合成了具有這些基本特性的兩種添加劑三氟(氰基)硼酸鋰(LiBF3CN或LCTFB)和氰基三(2,2,2-三氟乙基)硼酸鋰(LiB(OCH)2CF3)3CN,或LCTFEB),并與LiBF4進行比較。盡管具有相似的結構(圖1),但電池性能和SEI特性都因添加劑的選擇而異。循環 Li/Li 對稱電池,頻率為 1 mA/cm2LCTFB電解質的穩定循環超過650小時,LCTFEB電解質的循環穩定超過500小時,但LiBF4的電池失效在400小時內發生,對于不含添加劑的標準電解質,在200小時內發生。為了分析更實用的鋰金屬電池中的電解質,構建了Li/NCM523電池并在0.5C下循環。300次循環后,電池用LiBF4循環容量保持率為83.1%,而用LCTFB電解質循環的電池的容量保持率為98.4%,電壓遲滯顯著降低(圖5d)。相比之下,使用無添加劑電解質循環的電池在84次循環后具有2.236%的容量保持率,而使用LCTFEB電解質循環的電池在91次循環后具有0.500%的容量保持性(圖5e)。添加劑上配體的選擇也會影響沉積的鋰和隨后形成的SEI的形態。在標準電解液中,不含添加劑,含LiBF4,苔蘚狀針狀枝晶沉積在表面上,而LCTFB和LCTFEB電解質具有更光滑的表面。
此外,LCTFEB電解質循環會產生大而笨重的鋰顆粒(>1μm)。電極的冷凍透射電鏡分析表明,SEI 的厚度也因電解質而異,不含添加劑的標準電解質和 LiBF4 的鋰厚度為 30、40、20 和 10 nm、LCTFB 和 LCTFEB(圖 5b,c)。用無添加劑電解質循環的負極上的厚SEI表明對鋰離子擴散具有很高的抵抗力,導致鋰縱向生長并產生用標準電解質和LiBF4觀察到的針狀枝晶結構電解質。由于鋰離子通過薄SEI的擴散相對較快,因此鋰顆粒的直徑能夠增長,導致在與LCTFB和LCTFEB電解質循環時形成更大的顆粒。形成的SEI的化學成分也因電解質而異,因此,在具有相似結構特征的合成添加劑上選擇配體可以極大地影響SEI的形貌和組成,并導致鋰金屬負極性能的提高。
總結與展望
1. 協同鹽和添加劑組合,實現多個SEI設計目標
SEI中的柔韌性和導電性都可改善鋰金屬負極性能,其形成的表面膜能夠保持穩定并防止電解質連續分解。但是,大多數組分僅解決其中一個目標。如果可以選擇雙鹽來提供一些增加的離子電導率而不會顯著消耗鋰離子載體,除了優先還原以形成聚合物SEI的添加劑之外,那么這兩個方面都可以實現。因此,添加劑和鹽應以不同的比例和品種以組合方式進行測試,以潛在地實現任何協同效應。
2. 金屬鋰預處理優先產生SEI
如果需要某些不一定溶于碳酸酯電解質的組分,則鋰金屬負極表面的預處理可以幫助將這些組分摻入SEI中而不會原位形成。例如,在組裝電池之前表面的聚合物涂層與電解質中的高導電添加劑相結合,可以使鋰金屬繼續電鍍和剝離,同時防止形成死鋰。
3. 改性電解質與高壓正極的相容性
改性碳酸酯電解質(而不是另一種溶劑類型)的主要優點是碳酸酯在高壓正極上的穩定性。因此,任何新型添加劑或獨特的鹽混合物都應在高電位下進行測試,以查看是否觀察到任何對整個電池有害的降解。
4. SEI的結構控制
雖然較少討論,但SEI和沉積鋰顆粒的納米結構和形態對于持續的鋰金屬電鍍/剝離至關重要。此外,如果可以設計出促進納米結構沉積和控制SEI生長的預鋰化涂層或添加劑,則鋰金屬負極性能可能會有所改善。
5. 以工業規模測試電解液
這里討論的大多數電解質研究都是在實驗室中小規模(紐扣電池)完成的。在紐扣電池中,電解質量和鋰源相對不受限制。然而,在考慮未來在工業和商業規模上的應用時,由于電池組的生產成本和能量密度,需要考慮電解液的數量和鋰箔的厚度。因此,應在軟包電池和棱柱形電池上進一步測試有希望的改性電解質,以確保所有降解產物在更大范圍內都是安全的,并且不會造成額外的危害。此外,電解液應符合所有工業標準,包括最小電解液量和電極負載速率。
審核編輯:劉清
-
鋰離子電池
+關注
關注
85文章
3238瀏覽量
77686 -
鋰電池
+關注
關注
260文章
8098瀏覽量
169943 -
電解質
+關注
關注
6文章
810瀏覽量
20049 -
電解液
+關注
關注
10文章
847瀏覽量
23092
原文標題:羅德島大學Brett L. Lucht教授最新ACS Energy Lett.:優化碳酸酯基電解液用于鋰負極
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
水系電解液寬電壓窗口設計助力超長壽命水系鈉離子電池
離子液體添加劑用于高壓無負極鋰金屬電池

貼片電解電容正負極判斷方法
鎳氫電池的電解液是什么
新宙邦擬在美國投建10萬噸/年電解液項目
新宙邦美國路易斯安那州碳酸酯溶劑和鋰離子電池電解液項目啟動
用于延長高壓高Ni三元鋰金屬電池壽命的無氟醚基電解液

液位傳感器監測鉛酸電池電解液液位

非質子型弱配位電解液實現無腐蝕超薄鋅金屬電池

弱溶劑化少層碳界面實現硬碳負極的高首效和穩定循環







 工商網監
工商網監
評論