 高壓、快動力學鈉金屬電池中的競爭配位
高壓、快動力學鈉金屬電池中的競爭配位
研究背景
鈉基電池因其與鋰基電池相似的儲能機制以及鈉資源分布廣、成本低等特點,被認為是鋰基電池的互補技術。為了實現更高的能量密度,毫無疑問,鈉金屬是鈉基電池的最終負極,因為它具有高理論比容量(1166 mAh g–1 )和低氧化還原電位(?2.714 V vs SHE)。然而,鈉金屬負極面臨的問題與鋰金屬負極相似甚至更嚴重。鈉金屬反應性更高,與電解液發生更嚴重副反應,形成化學/電化學穩定性較差的脆弱固體電解質界面(SEI)。在隨后的重復剝離/電鍍過程中,SEI溶解并破裂,進一步導致鈉枝晶的生長和死鈉的形成。這種不穩定的界面結構導致活性鈉的不可逆損失,嚴重惡化了鈉金屬電池(SMBs)的安全性和循環穩定性。此外,鈉的氧化還原電位 (+0.3 V vs Li) 高于鋰,這意味著在全電池相同的充電截止電壓下,SMBs 中使用的電解液需要更高的氧化電位,這也是高壓 SMBs 實現高能量密度的嚴峻挑戰。盡管在構建合適的SEI方面報道了大量研究性工作,但迄今為止,電解液溶劑化結構中陽離子-溶劑配位和陽離子-陰離子配位對SEI結構的影響很少被研究和報道。事實上,SEI的組成和分布對溶劑化結構中陽離子-溶劑和陽離子-陰離子之間的競爭配位高度敏感。
研究簡介
近日,中國科學院金屬研究所的楊慧聰、李峰團隊基于焓變影響配位的本質,通過調控鈉離子競爭配位的策略設計了一種具有穩定電極-電解液界面、高氧化穩定性、快速鈉離子輸運動力學等綜合性能優異的新型電解液體系。通過對溶劑氟取代后,鈉離子-溶劑的結合較弱,形成鈉離子-溶劑配位結構比形成鈉離子-陰離子配位結構導致的焓釋放更少。此外,這種低焓的配位結構可以提高競爭配位平衡對鹽濃度的敏感性,即使在電解液中鹽濃度相對較低的情況下,原始的競爭配位平衡也會發生變化,使更多的陰離子在溶劑化鞘中進行配位。因此,該電解液中Na||Cu電池具有98.0%的高庫倫效率,Na||NVPF電池具有優異的循環穩定性,在0.5/1C下500次循環后平均CE 99.4/99.6%,容量保持率為94.3/94.1%。該文章以 “Competitive Coordination of Sodium Ions for High-Voltage Sodium Metal Batteries with Fast Reaction Speed”為題發表在Journal of the American Chemical Society上。
圖文導讀
1. 溶劑設計對鈉離子的競爭性協調調控
如圖1a所示,與DEC和EC相比,ETC和FEC中氧原子周圍的負電荷較少,導致Na+與溶劑之間的配位較弱(圖1b)。與純溶劑相比,Na+-溶劑的HOMO能級較低,且Na+- PF6-負能級最大(圖1c),說明ETC和FEC溶劑分子的加入以及同時增強的Na+- PF6-配位可以獲得更高的電解液氧化穩定性 (圖1d)。拉曼光譜表明隨著氟化水平的增加,1 M NaPF6-DEC/FEC(DF電解液)和1 M NaPF6-ETC/FEC(EF電解液)的PF6-峰分別藍移至739.7 cm-1和741.5 cm-1,表明陰離子參與溶劑化結構增加。還考察了不同氟化水平的結構對鹽濃度的敏感性。當鈉鹽濃度從1 M增加到1.3 M時(圖1e), DEC/EC電解液(DE電解液)中沒有觀察到PF6-陰離子的藍移或紅移,保持了原有的溶劑化結構。而1.3 M NaPF6-ETC/FEC中PF6-峰呈現藍移。由于溶劑化結構變化對鹽濃度的敏感性,EF電解液中鹽濃度的輕微增加可以進一步促進電極上陰離子衍生的界面化學反應。傅里葉變換紅外光譜(FTIR)中,Na+-溶劑與游離溶劑的比例隨著?F基團的存在而降低:Na+-ETC(12.3%)<Na+-DEC(22.4%),Na+-FEC(72.9%)<Na+-EC(76.8%)。因此兩種氟化溶劑的EF電解液表現出優化的溶劑化結構,總體Na+-溶劑配位降低, Na+-陰離子配位增加,圖1f中 19F核磁共振(NMR)光譜的化學位移變化進一步驗證了這一點。
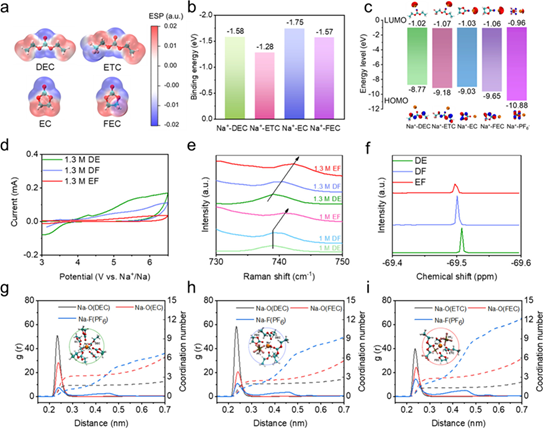
圖1:(a)不同溶劑分子的靜電勢(ESP)。(b) Na+與不同溶劑的結合能。(c) Na+-溶劑和Na+-陰離子的HOMO和LUMO能級。(d) 1.3 M電解液的氧化穩定性的CV測試。(e)不同電解液體系中Na+-PF6-的配位。(f) DE、DF和EF電解液的19F NMR譜。(g) DE, (h) DF和(i) EF電解液的徑向分布函數和代表性溶劑化結構(插圖)。
為了深入了解溶劑氟化對鈉離子競爭配位的影響,進行了經典分子動力學(MD)模擬。當EC被FEC取代時,Na+-FEC表現出比Na+-EC更長的相互作用距離和更低的總Na+-溶劑配位數(圖1h)。當DEC進一步被ETC取代時, Na+-溶劑的總配位數進一步降低,Na-F(PF6–)和Na-O(FEC)的配位數增加(圖1i)。MD模擬結果與拉曼和FTIR光譜結果一致,證實了通過溶劑設計可以調節鈉離子的競爭配位,從而實現富含陰離子的溶劑化結構。
2. 鈉金屬負極的界面分析
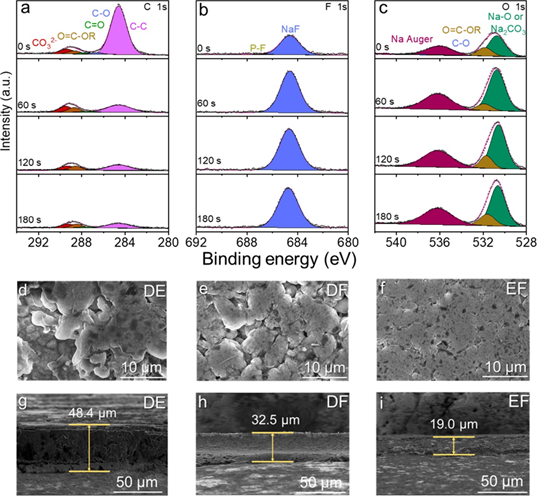
圖2.EF電解液在0.5 mA cm–2下循環20圈后的SEI的(a)C 1s、(b)F 1s和(c)O 1s XPS 光譜。在0.5 mA cm –2下(d) DE、(e) DF 和 (f) EF 電解液中沉積鈉形態的 SEM 圖像。(g-i)在0.5 mA cm–2 下用不同電解液循環后SEI的橫截面SEM圖像。
采用X射線光電子能譜(XPS)分析不同電解液中SEI的組分,進一步揭示鈉離子競爭配位變化與SEI的密切關系。EF電解液中C的原子含量降至最低(7%),F的原子含量增加到12%(圖2a)。同時,F 1s譜沒有檢測到相關的副產物CFx 和P-F物種(圖2b),這是由于通過增強Na+-PF6–和Na+-FEC配位,導致FEC和PF6–更徹底地分解。此外,ROCO2Na在SEI內層可能轉化為Na2O或Na2CO3,從而形成更多無機組分Na2O(圖2c)。由于DE和DF電解液中 Na+-溶劑配位較強,更多的溶劑在Na/電解液界面被還原和不完全分解,從而產生有機主導且更厚的SEI。相反,結合EF電解液中Na+-溶劑配位的降低和Na+-陰離子配位的增加,降低了溶劑分解,獲得了富含無機物且堅固的SEI,有利于抑制鈉枝晶的形成,加速Na+在SEI中的遷移,從而實現鈉的均勻沉積。通過掃描電子顯微鏡(SEM)觀察銅箔上鈉金屬電沉積的形貌。在EF電解液中觀察到沉積鈉金屬更加平坦均勻,沒有枝晶或死鈉(圖2f),在20次循環后,具有更低的厚度(19.0μm,圖2i)。這些事實證實,調節鈉離子的競爭配位以獲得修飾的溶劑化結構可以形成高度穩定的SEI,以抑制枝晶和副反應。
3. 鈉金屬負極的沉積/剝離可逆性
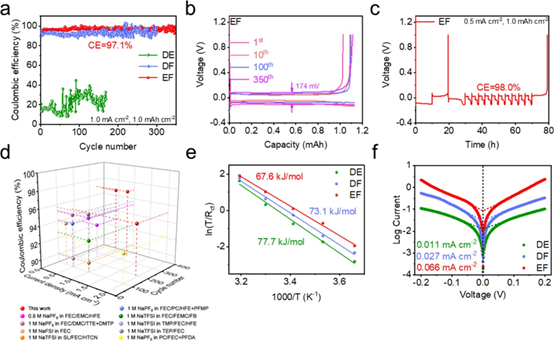
圖3.(a)Na||Cu電池在1.0 mA cm–2 –1.0 mAh cm–2下的CE和(b)EF電解液的電壓曲線。(c)采用改進方法測試EF電解液在 0.5 mA cm–2 –1.0 mAh cm–2 的CE。(d)與已報道的酯類電解液中Na||Cu電池的CE對比。(e)不同電解液的去溶劑化活化能和(f)交換電流密度。
組裝Na||Cu電池以評估鈉金屬負極的可逆性和電鍍/剝離的穩定性(圖3a) DE 電解液在 100 次循環中顯示出 21.7% 的低平均 CE。在FEC存在的情況下,DF電解液的平均CE明顯更高,約為92.7%,表明FEC在循環過程中對SEI有積極影響。此外,EF電解液的平均CE達到97.1%,極化電壓較低(第350次循環為174 mV;圖3b)。采用改進后的CE測試方法更準確地評估鈉電鍍/剝離的可逆性,從而獲得了98.0%的高CE(圖3c和圖3d)。EF電解液的對稱電池在0.5 mA cm–2 –1.0 mAh cm–2 和1.0 mA cm–2 –1.0 mAh cm–2下可以穩定循環超過1400小時和1000小時。此外,EF電解液對Na+的去溶劑化具有最低的活化能,證實了快速去溶劑動力學(圖3e)。此外,隨著電解液溶劑氟化水平的增加,可以觀察到交換電流密度從0.011 mA cm–2 (DE)逐漸增加到0.027 mA cm–2(DF)和0.066 mA cm–2(EF),如圖3f所示。結果表明我們通過溶劑設計來調節鈉離子競爭配位的策略成功地優化了溶劑化結構,從而產生了穩固的SEI、優異的循環穩定性和快速的電化學動力學。
4. Na||NVPF電池的電化學性能
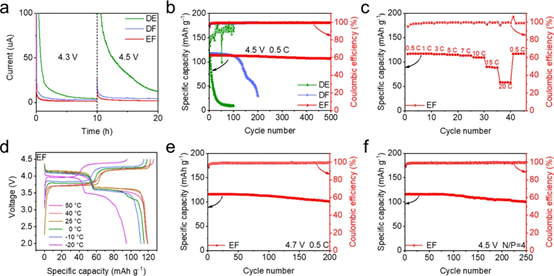
圖4.(a)Na||NVPF電池在4.3和4.5V恒定電壓下的CA曲線。(b)4.5V、0.5C下Na||NVPF電池的循環測試。(c)4.5V下EF電解液的倍率性能測試。(d)?20至50°C,4.5V、0.5C下EF電解液的充放電曲線。(e)4.7V、0.5C下EF電解液的Na||NVPF電池循環穩定性。(f)4.5V、0.5C下EF電解液Na||NVPF全電池測試(N/P=4:1)。
鑒于鈉金屬負極在EF電解液中的優異性能,我們組裝了Na||NVPF電池,用于評估所設計的電解液在高電壓(≥4.3 V)的全電池中的實用性。首先如圖4a所示,當電壓保持在4.3 V時,與DE(4.4 μA)和DF(3.0 μA)電解液相比,EF電解液表現出最小的漏電流(1.3 μA)和最短達到穩態時間,并且在4.5 V時觀察到相同的趨勢。這些結果證實了EF電解液在NVPF陰極上具有最高的氧化穩定性和最小的副反應,與DFT計算和CV測試的結果一致(圖1c,d)。在 4.3 V 和 0.5 C 下采用EF電解液的Na||NVPF電池具有低電壓極化和出色的循環穩定性,在500次循環中容量保持率為95.7%。4.5V下,采用EF電解液的Na||NVPF電池平均CE超過99.4/99.6%,在0.5/1C下500次循環后容量保持率為94.3/94.1%(圖4b)。此外,由于快速的鈉離子傳輸動力學, EF電解液表現出優異的倍率性能和寬溫性能(圖4c和圖4d)。即使在 4.7 V 的超高截止電壓下, EF電解液的Na||NVPF電池也可以實現超過200圈的穩定循環(圖4e)。此外,組裝了N/P比為4:1的 Na||NVPF全電池,在 250 次循環后仍具有初始容量的 87.1%(圖4f)。
5. 鈉離子競爭配位的本質

圖5.(a、b)Na+-溶劑和Na+-陰離子之間競爭配位平衡的示意圖。Na+-溶劑和Na+-陰離子結合能的相對變化使溶劑化結構的競爭配位平衡從Na+-溶劑向增加的Na+-陰離子轉變,這是由焓變決定的。
實際上,SMBs電解液中不同溶劑化結構的比例是由陽離子-溶劑和陽離子-陰離子之間的競爭配位決定的。而這種競爭配位平衡本質上是由形成不同溶劑化結構時的焓變決定的。Na+-溶劑或Na+-陰離子配位結構最終形成取決于哪種配位結構可以實現最小的吉布斯自由能。電解液的吉布斯自由能隨Na+-溶劑和Na+-陰離子的含量而變化,即Na+-溶劑或Na+-陰離子配位結構形成時釋放的焓變,而焓變由結合能決定。如圖6a所示,當使用EC和DEC溶劑時,Na+-溶劑的高結合能通過形成Na+-溶劑配位結構而比形成Na+-陰離子配位結構引起更多的焓釋放。因此,DE電解液中的溶劑化結構以Na+-溶劑配位為主,以實現最小的吉布斯自由能。然而,在溶劑的氟取代下,Na+-溶劑較弱的結合通過形成Na+-溶劑配位結構比形成Na+-陰離子配位結構導致的焓釋放更少(圖5b)。此外,這種釋放焓較少的Na+-溶劑配位結構可以提高競爭配位平衡對鹽濃度的敏感性。即使EF電解液在相對較低的鹽濃度下,原始的競爭配位平衡也會發生變化,使更多的陰離子在溶劑化鞘中配位。這種調控陽離子競爭配位平衡的策略不僅適用于鈉金屬電池的電解液設計,而且可以擴展到其他具有相同熱力學本質的電池體系中的電解液溶劑化結構設計。
總結和展望
綜上所述,本文基于焓變影響平衡的性質,設計了一種新電解液體系,實現了具有快速電化學動力學和優異循環穩定性的高壓鈉金屬電池。通過調節鈉離子的競爭配位,獲得了具有顯著陰離子配位的溶劑化結構,從而形成富含無機物的電極-電解液界面、高氧化穩定性和快速去溶劑化動力學。Na||NVPF 電池在 4.5 V 的高截止電壓下具有出色的倍率能力、寬工作溫度范圍和長期循環穩定性。所提出的通過調節陽離子的競爭配位來改變溶劑化結構的策略,為實現穩定的高能量密度鈉金屬電池提供了新的方向,并且可以通過控制溶劑化結構的焓變進一步擴展到其他電池體系。
審核編輯:劉清
-
鋰電池
+關注
關注
260文章
8098瀏覽量
169940 -
電解液
+關注
關注
10文章
847瀏覽量
23092 -
傅里葉變換
+關注
關注
6文章
441瀏覽量
42592 -
鈉電池
+關注
關注
0文章
73瀏覽量
10234 -
拉曼光譜
+關注
關注
0文章
83瀏覽量
2738
原文標題:中國科學院金屬研究所楊慧聰、李峰團隊JACS:高壓、快動力學鈉金屬電池中的競爭配位
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
多功能高熵合金納米層實現長壽命無負極鈉金屬電池

使用Phase Lab鎳基動力學數據庫計算多組分合金的成分分布曲線
“本源悟空”超導量子計算機助力大規模流體動力學量子計算

Simcenter STAR-CCM+車輛外部空氣動力學特性——通過快速準確的CFD仿真加速空氣動力學創新

圓滿收官| Aigtek參展第二屆波動力學前沿與應用學術會議載譽歸來!











 工商網監
工商網監
評論