


1、導讀

清華大學深圳國際研究生院康飛宇(本刊主編)、賀艷兵(本刊執行主編)、柳明(本刊青年編委)、侯廷政(本刊青年編委)團隊闡明了 BaTiO3鐵電填料誘導 LiFSI解離的機制。通過引入不同極化態的 BaTiO3(BTO)填料,揭示了自發極化影響下 LiFSI 的解離機制。具有氧空位缺陷的四方 BTO3?x顯著提升了 LiFSI 在 PVDF 復合固態電解質中的解離度,生成了 72% 的高濃度自由Li+,從而提高了離子電導率,達到 8.4×10?4S cm?1。該文章以 “Dissociation mechanism of lithium salt by BaTiO3 with spontaneous polarization”為題發表在Energy & Environmental Science上。第一作者是郭少柯和譚神冬。
2、研究背景
固態電解質搭配鋰金屬負極和高鎳正極,能夠實現高安全性和可達400 Wh/kg的能量密度。這使得它們成為電動車(EVs)和能量儲存系統(ESSs)的理想選擇。在這個過程中,固態聚合物電解質(SPE)由于其柔韌性和易加工性的特點,成為比固態無機電解質更有前景的選擇。然而,傳統聚合物基電解質在室溫下的高結晶性阻礙了鋰離子(Li+)的遷移,導致室溫下難以運行。值得注意的是,在聚偏氟乙烯(PVDF)基聚合物電解質中,由于殘留的N,N-二甲基甲酰胺(DMF)參與到鋰離子的溶劑化結構中,削弱了鋰離子與PVDF鏈之間的結合作用,因此,形成的[Li(DMF)x]+顯著增強了PVDF基電解質在室溫下的離子傳輸性能。但是,在PVDF電解質中,大多數鋰離子以接觸離子對(CIP)和聚集團簇(AGGs)的形式存在,而不是自由Li+。這種獨特的溶劑結構為進一步提高鋰離子傳輸效率造成了障礙。最近的研究發現,具有鐵電特性的功能填料能夠促使鋰鹽(LiFSI)解離,從而顯著提高離子電導率。然而,要完全理解其內在機理卻很困難,這阻礙了電解質性能的進一步提高。
3、工作要點
在這項研究中,作者選擇了立方相鈦酸鋇(C-BTO)和四方相鈦酸(T-BTO)作為代表性模型來探討LiFSI在自發極化影響下的解離機制(圖1 a,1b)。研究表明, T-BTO的偶極矩方向可以在電池的外部電場作用下略微偏轉,以增強極化效果。沿著自發極化方向的T-BTO {001} 面展現出更加顯著的LiFSI解離能力,并且能夠吸附FSI?陰離子,這一效果通過引入氧空位(OV)缺陷得到進一步放大(圖1c)。
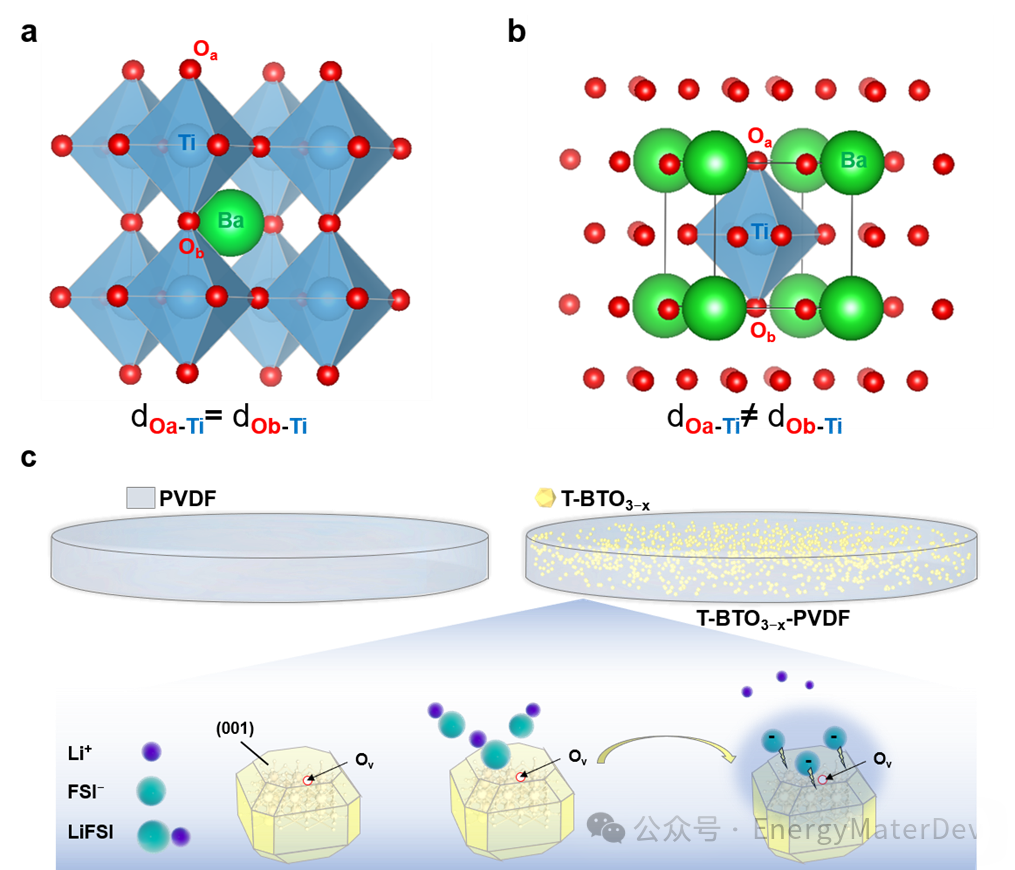
圖 1 填料的晶體結構以及填料與 LiFSI 之間的相互作用過程示意圖。a) C-BTO 的晶體結構,b) T-BTO 的晶體結構;c) T-BTO3?x的{001}面上 LiFSI 的解離和吸附過程示意圖。
通過透射電子顯微鏡(TEM)圖像,可觀察到0.288nm和0.396nm的晶面間距,分別與C-BTO的(110)面和T-BTO的(100)面相吻合(圖2a, 2b)。壓電顯微鏡(PFM)展示了T-BTO的類矩形磁滯回線,表明其具有自發極化性質,而C-BTO的磁滯回線不明顯(見圖2c, S3)。為模擬電池環境中的實際極化情況,作者用薄膜鐵電分析儀施加了與電池相當的外部電場(4V,80μm)。如圖2d所示,T-BTO的磁滯回線未重疊并閉合,表明T-BTO中偶極子方向略微偏轉,有助于極化的增強。為進一步增強自發極化,通過控制在H2-Ar混合氣氛中燒結時間引入了氧空位,從而形成具有不同氧空位濃度的T-BTO3?x。T-BTO3?x的電子自旋共振(ESR)表明隨著燒結時間延長, T-BTO3?x中氧空位濃度相應增加(圖2e)。在沒有LiFSI的T-BTO-PVDF薄膜中,其介電常數(εr)在10Hz時為46,在燒結6小時后,隨著氧空位濃度的增加,εr最大可達59(圖2f)。
此外,電子能量損失譜(EELS)提供了O-K(O1s 2p)微觀結構信息,估計T-BTO3?x的表面氧濃度可以表示為T-BTO2.87(圖2g)。研究中利用XRD的Rietveld精修揭示了T-BTO2.87的結構變化。在T-BTO和T-BTO2.87中的TiO6八面體中,dOa-Ti的距離從2.155?增加到2.197?,而dOb-Ti的距離從1.883?減少到1.838?(見圖2h,2i,S5a,S5b)。dOa-Ti和dOb-Ti之間的距離差從0.272?擴大到0.359?。這表明由于氧空位的存在,Ti4+從TiO6八面體中心的電荷偏移程度顯著增強,相應增加了自發極化。
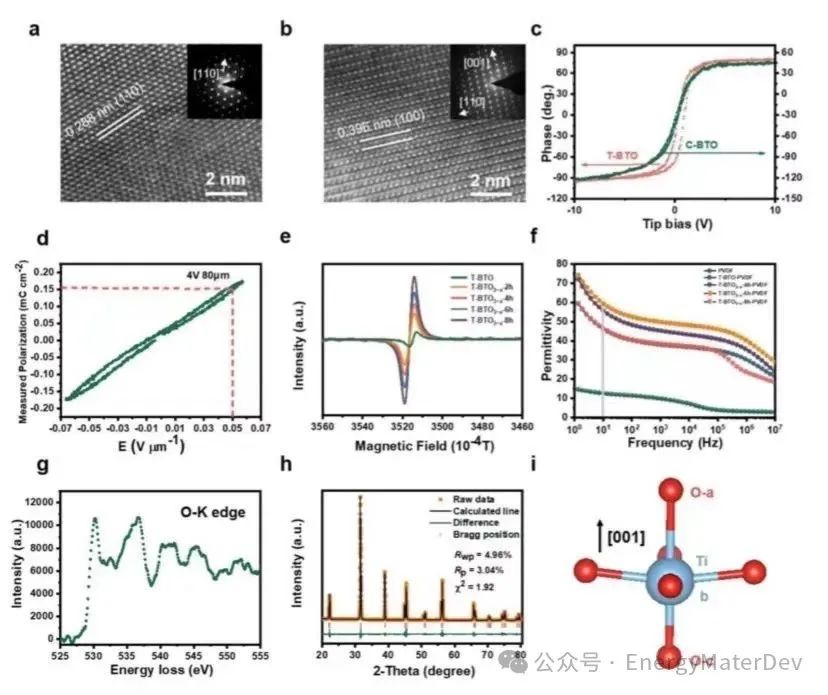
圖2 C-BTO、T-BTO 和 T-BTO3?x的特性。a) C-BTO 顆粒和 b) T-BTO 顆粒的 TEM;c) 通過 PFM 分析獲得的 C-BTO-PVDF 和 T-BTO-PVDF 的電滯回線;d) T-BTO 在電池電場下的鐵電回線;e) 電子自旋共振,用于半定量氧缺陷濃度;f) PVDF、C-BTO-PVDF、C-BTO3?x-PVDF、T-BTO-PVDF和T-BTO3?x-PVDF的介電常數;g) T-BTO3?x O-K的電子能量損失圖譜;h) T-BTO3?x的XRD Rietveld精修結果;i) T-BTO3?x的Ti4+偏移圖。
為了進一步說明T-BTO與FSI?之間的相互作用,作者使用zeta電位在不同LiFSI濃度的DMF溶液中測試了填料的電荷狀態。如圖3f所示,在沒有LiFSI的DMF中,C-BTO和T-BTO的zeta電位接近0。隨著LiFSI濃度的增加,T-BTO的zeta電位顯著下降,而C-BTO的zeta電位只輕微下降。由于FSI?是系統中唯一的負電荷粒子,這種差異表明,具有不同暴露晶面的T-BTO比C-BTO更有效地吸附FSI?(圖3g)。通過繪制Wulff圖,獲得了受表面能影響的不同暴露晶面的分布(圖3h)。因為C-BTO的晶體結構可以通過沿<001>方向拉伸扭曲為T-BTO的結構,因此它們的主要性質差異發生在{001}晶面上。通過使用DFT模擬C-BTO和T-BTO {001}晶面與FSI?的相互作用(圖3h),可以觀察到T-BTO {001}晶面顯示出比T-BTO {100}和C-BTO {001}晶面更強的吸附能力(?0.360eV)。
因此,T-BTO {001}晶面在吸附FSI?中起著關鍵作用。為了更深入理解相互作用機制,作者進一步通過DFT模擬了鋰鹽的離解過程。如圖3i所示,在體相中,Li+傾向于與FSI?的兩個O形成雙齒結構。然而,當LiFSI靠近T-BTO的{001}晶面時,FSI?的O傾向于與T-BTO的Ti結合,形成表面吸附的FSI?,從而使Li+從FSI?中解離。在表面引入氧空位進一步增強了這一現象。T-BTO3?x的zeta電位(?4.03mV)比T-BTO(?3.72mV)更負,且Li+的遷移數從PVDF的0.18和T-BTO-PVDF的0.29增加到T-BTO-PVDF3?x的0.52。
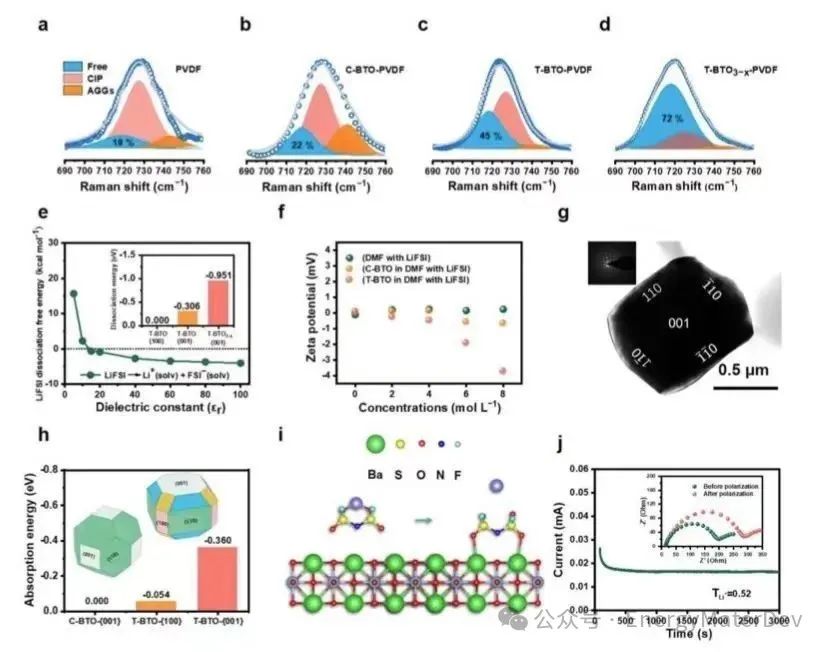
圖3 LiFSI 的解離機理。a–d)不同填料電解質的拉曼位移;e)LiFSI 解離自由能與介電常數關系的理論計算;f)C-BTO和T-BTO 在不同鋰鹽濃度的 DMF 中的 Zeta電位;g)Z≈[001]方向的明場TEM和相應的選區電子衍射;h)不同表面對FSI-的吸附能計算;i) LiFSI 解離過程說明圖;j) Li||T-BTO3?x-PVDF||Li 對稱電池的電流–時間曲線和Li+遷移數。
納米紅外表明,PVDF 電解質中的DMF分布不均勻(圖4a, 4b),而在T-BTO3?x-PVDF電解質中,DMF圍繞填料均勻分布(圖4c, 4d)。DMF的均勻分布有效地增強了Li+的傳輸,并減少了由于DMF的聚集而導致的界面副反應。此外,引入T-BTO3?x填料后,激活能(Ea)從0.3eV顯著下降到0.179eV(圖4e,S12)。因此,在25℃時,T-BTO3?x-PVDF的離子導電率達到8.4×10?4 S cm?1,而PVDF為4.2×10?4 S cm?1,C-BTO-PVDF為3.3×10?4 S cm?1,T-BTO-PVDF為6.5×10?4 S cm?1(圖4f,S13)。
電化學測試揭示了T-BTO3?x-PVDF電解質在鋰金屬負極界面的優勢。Tafel曲線顯示,Li||T-BTO3?x-PVDF||Li電池的交換電流密度(j0=0.63 mA cm?2)明顯高于Li||T-BTO-PVDF||Li電池(j0=0.31 mA cm?2)和Li||PVDF||Li電池(j0=0.05 mA cm?2)(圖4g)。這表明在鋰和電解質之間形成了更具動力學上有利的電荷轉移界面。此外,填料可以將T-BTO3?x-PVDF的電化學窗口從3.7 V增加到4.4 V,這是由于其與高鎳正極的優異兼容性(圖S14)。隨著T-BTO3?x的引入,T-BTO3?x-PVDF的拉伸強度從PVDF1.99MPa增加到2.45MPa(圖S15)。
Li||T-BTO3?x-PVDF||Li對稱電池的臨界電流密度(CCD)可以達到2.2 mA cm?2,超過了匹配PVDF(0.6 mA cm?2)、C-BTO-PVDF(0.9 mA cm?2)和T-BTO-PVDF(1.4 mA cm?2)的電池(圖4h)。此外,Li||T-BTO3?x-PVDF||Li對稱電池在0.1 mA cm?2下顯示了超過2000小時的長循環壽命)。值得注意的是,在0.5 mA cm?2的電流密度下,T-BTO3?x-PVDF的優勢更加顯著,表現出長達700小時的穩定循環時間,而其他電解質則不到210小時(圖4i)。即使在電流密度為1 mA cm?2時,Li||T-BTO3?x-PVDF||Li對稱電池也能保持穩定的循環超過376小時(圖4j)。這些結果證實了T-BTO3?x可以極大地提高鋰負極與T-BTO3?x-PVDF電解質之間的界面穩定性和動力學特性。
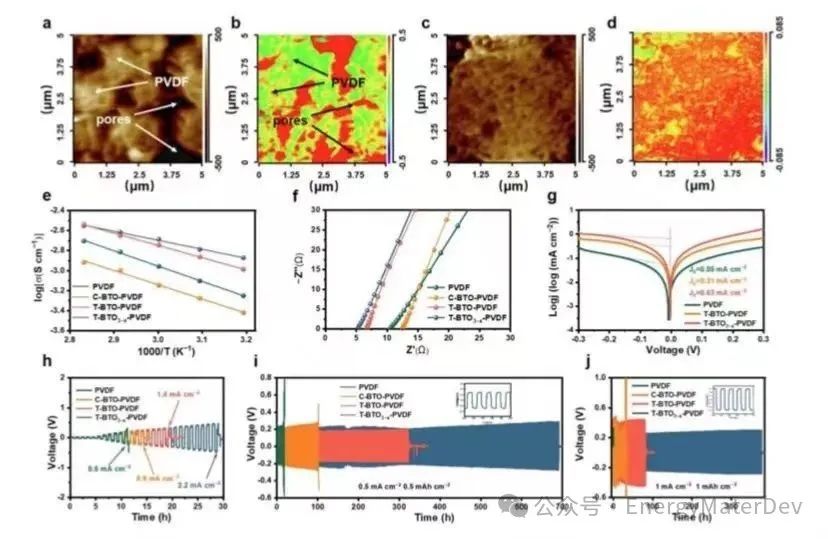
圖4 T-BTO3?x-PVDF 電解質的特性。a) PVDF、c) T-BTO3?x-PVDF 的原子力顯微鏡圖像,以及 b) PVDF 和 d) T-BTO3?x-PVDF相應的納米紅外圖像(DMF 的 C=O 振動);e) PVDF 基電解質的Arrhenius圖;f)鋼鋼(SS)||SS 電池的Nyquist阻抗譜;g) 不同電解質的Li||Li對稱電池的塔菲爾圖;h) 不同電解質的Li||Li對稱電池的極限電流密度和不同電解質的Li||Li對稱電池在i)0.5 mA cm?2-0.5 mAh cm?2 j)1 mA cm?2-1 mAh cm?2下的長循環。
圖5 NCM811||Li電池的電化學性能和界面表征;a) 25 ℃時,NCM811||Li電池的倍率性能;b) 0.5 C和c) 1 C 下的循環性能;d) PVDF和g) T-BTO3?x-PVDF的正極XPS圖譜;循環后的NCM811顆粒與e, f) PVDF 和 h, i) T-BTO3?x-PVDF匹配的TEM圖像。j, l) PVDF 電解質和k, m) T-BTO3?x-PVDF 電解質循環后鋰金屬表面Li2OH+和Li3CO3+的TOF-SIMS 3D重構。
電化學測試揭示了T-BTO3?x-PVDF電解質在鋰金屬負極界面的優勢。Tafel曲線顯示,Li||T-BTO3?x-PVDF||Li電池的交換電流密度(j0=0.63 mA cm?2)明顯高于Li||T-BTO-PVDF||Li電池(j0=0.31 mA cm?2)和Li||PVDF||Li電池(j0=0.05 mA cm?2)(圖4g)。這表明在鋰和電解質之間形成了更具動力學上有利的電荷轉移界面。此外,填料可以將T-BTO3?x-PVDF的電化學窗口從3.7 V增加到4.4 V,這是由于其與高鎳正極的優異兼容性匹配(圖S14)。此外,隨著T-BTO3?x的引入,T-BTO3?x-PVDF的拉伸強度從PVDF(1.99 MPa)增加到2.45 MPa(圖S15)。
進行了Li||T-BTO3?x-PVDF||Li對稱電池測試以揭示BTO3?x的特殊影響。Li||T-BTO3?x-PVDF||Li對稱電池的臨界電流密度(CCD)可以達到2.2 mA cm?2,超過了使用PVDF(0.6 mA cm?2)、C-BTO-PVDF(0.9 mA cm?2)和T-BTO-PVDF(1.4 mA cm?2)的電池(圖4h)。此外,Li||T-BTO3?x-PVDF||Li對稱電池在0.1 mA cm?2下展示了超過2000小時的長循環壽命,而與PVDF、C-BTO-PVDF和T-BTO-PVDF相比分別為390小時、660小時和941小時(圖S16)。值得注意的是,在0.5 mA cm?2的電流密度下,T-BTO3?x-PVDF的優勢更加顯著,表現出穩定的循環時間長達700小時,而其他電解質則不到210小時(圖4i)。即使在臨界電流密度為1 mA cm?2時,Li||T-BTO3?x-PVDF||Li對稱電池也能保持穩定的循環超過376小時(圖4j)。這些結果證實了T-BTO3?x可以極大地提高鋰陽極與T-BTO3?x-PVDF電解質之間的界面穩定性和動力學特性。
速率性能方面,0.2 C 下,NCM811||PVDF||Li(179.7 mA h g?1)最初的放電比容量略低于NCM811||T-BTO3?x-PVDF||Li(185.8 mA h g?1)。隨著速率的增加,NCM811||PVDF||Li的放電比容量(92.6 mA h g?1)遠遠落后于NCM811||T-BTO3?x-PVDF||Li(107.7 mA h g?1),表明BTO3?x(圖5a)進一步改善了Li+的傳輸。當以0.5 C的速率循環時,NCM811||T-BTO-PVDF||Li和NCM811||T-BTO3?x-PVDF||Li在第250次循環時的容量保留率分別為88%和77%,而NCM811||PVDF||Li和NCM811||C-BTO-PVDF||Li電池分別在第86次和第170次時失效(圖5b)。這表明T-BTO3?x通過調整溶劑化結構進一步改善了Li+的傳輸。此外,NCM811||T-BTO3?x-PVDF||Li電池可以在1 C下穩定循環1400次,容量保留率為61%(圖5c)。與之形成鮮明對比的是,其他電池的循環次數不到900次,且放電容量更低。這些改進的性能歸因于通過增加自由Li+而引起的離子導電率增加。
NCM811顆粒在1C下循環100次后的電解質界面(CEI)照片顯示,與PVDF(7.2 nm)和T-BTO-PVDF(5.57 nm)相比,使用T-BTO3?x-PVDF的正極-電解質界面層更薄(2.6 nm)(圖5e, 5h和圖S19),巖鹽相(C位點)和混合相(B位點)的厚度僅約為6 nm(圖5i)。改善的溶劑化結構通過引入增強極化的填料減少了副反應,并增強了正極動力學。使用T-BTO3?x-PVDF的鋰表面明顯比使用PVDF和T-BTO-PVDF的鋰表面更光滑(圖S21)。循環后鋰金屬表面的飛行時間二次離子質譜(TOF-SIMS)的顯示,與PVDF電解質(圖5j, l)相比,使用T-BTO3?x-PVDF電解質的SEI中Li2OH+和Li3CO3+的數量明顯減少(圖5k, 5m),導致更穩定的界面。這些結果證明了T-BTO3?x的自發極化填料可以通過改善界面穩定性來增強電化學性能。
4、總結與展望
本工作重點是通過比較 C-BTO、T-BTO 和增強自發極化 T-BTO3?x來理解自發極化對 LiFSI 解離的影響。通過實驗和理論模擬,研究人員發現 LiFSI 的解離程度與 T-BTO 的自發極化程度之間存在正相關。通過引入 Wulff 構造,研究人員提出了理論解釋,證實了解離吸附機制中自發極化面的主導地位。這項研究為了解局部極化環境對溶劑化結構的影響奠定了堅實的理論基礎,并為先進的復合固態電解質的設計提出了一種思路。
審核編輯:劉清
-
DFT
+關注
關注
2文章
234瀏覽量
23309 -
PFM
+關注
關注
1文章
166瀏覽量
28959 -
固態電解質
+關注
關注
0文章
86瀏覽量
5603
原文標題:清華大學康飛宇、賀艷兵、柳明、侯廷政最新EES:揭示固態電解質中鋰鹽解離的重要機制!
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
鉭元素賦能LLZO固態電解質,破解氧化物固態電池產業化密碼
鉭元素賦能LLZO固態電解質,破解氧化物固態電池產業化密碼
高臨界電流密度固態電池單晶鋰的合成

超聲波焊接有利于解決固態電池的枝晶問題
清華大學:自由空間對硫化物固態電解質表面及內部裂紋處鋰沉積行為的影響
研究論文::乙烯碳酸酯助力聚合物電解質升級,提升高電壓鋰金屬電池性能

陳軍院士團隊最新Angew,聚合物電解質新突破

Li3MX6全固態鋰離子電池固體電解質材料
一種薄型層狀固態電解質的設計策略
半互穿網絡電解質用于高電壓鋰金屬電池
通過電荷分離型共價有機框架實現對鋰金屬電池固態電解質界面的精準調控
全固態鋰金屬電池的鋰陽極夾層設計
固態電池中復合鋰陽極上固體電解質界面的調控






 工商網監
工商網監

評論