


研究背景
離子電池(LIBS)因其可回收性、高能量和高功率密度而廣受贊譽,使其在能源儲存系統、便攜式電子設備和電動汽車等各種應用中非常受歡迎。然而,使用易燃液體電解質和商用聚合物分離器的安全風險對其廣泛應用造成嚴重限制。在這種情況下,采用固體電解質的全固態鋰電池為提高安全性提供了巨大的潛力。在不同的粒子中,硫化物的離子導電性是非常好的。此外,硫化物SES還具有機械健壯性等優點,有可能穩定鋰金屬陽極,從而使高能量密度SSLBS的發展能夠滿足電動汽車中高范圍的需求。但基于硫化物的全固態電池商業化仍面臨諸多挑戰,包括化學不穩定性(對潮濕空氣和極性有機溶劑)及低能量密度(因SE層過厚)。為提高穩定性,表面修飾方法(如LiF等)存在難以保持材料性質均一性及影響離子電導率的問題。元素摻雜是較實用方法,軟酸陽離子摻雜及陰離子部分替換可提高穩定性,合理摻雜還可改變離子輸運結構,形成穩定界面層并增強對鋰金屬負極的穩定性。
成果簡介
北京科技大學范麗珍教授團隊提出In和F共摻雜策略,制備出高離子電導率、溶劑與電化學穩定性的硫化物固體電解質,通過濕法制膜實現高能量密度全固態電池。其中 6% InF3摻雜(Li5.82P0.94In0.06S4.7Cl1.12F0.18,簡稱LPSCInF)具有高化學穩定性,組裝的對稱電池在室溫下具有2.5 mA cm?2的超高臨界電流密度。在1 mA cm?2的電流密度下,Li/LPSCInF/Li對稱電池能夠穩定循環超過1000個小時。此外,In和F共摻雜的硫化物SE對水分子和有機溶劑具有很高的化學穩定性。組裝的LiCoO2/LPSCInF膜/Li電池在0.1 C條件下經過500個周期后具有83.2%的長循環壽命和高倍率性能。FeS2復合正極及固態電解質膜構建的全固態鋰金屬電池具有410 Wh kg?1的超高能量密度和優異的循環性能(3.8 mAhcm?2)。
研究亮點
1.通過提出In和F共摻雜策略,制備出高離子電導率、溶劑與電化學穩定性的硫化物固體電解質,通過濕法制膜實現高能量密度全固態電池。
2.In和F共摻雜的硫化物SE對水分子和有機溶劑具有很高的化學穩定性。
3. 制備了由LPSCInF和PIB粘結劑構成的硫化物SE膜。組裝的全固態電池首圈放電比容量為135.7 mAh g?1,初始庫侖效率為86.2%,經過500圈循環后容量保持率仍然高達83.2%。
圖文導讀
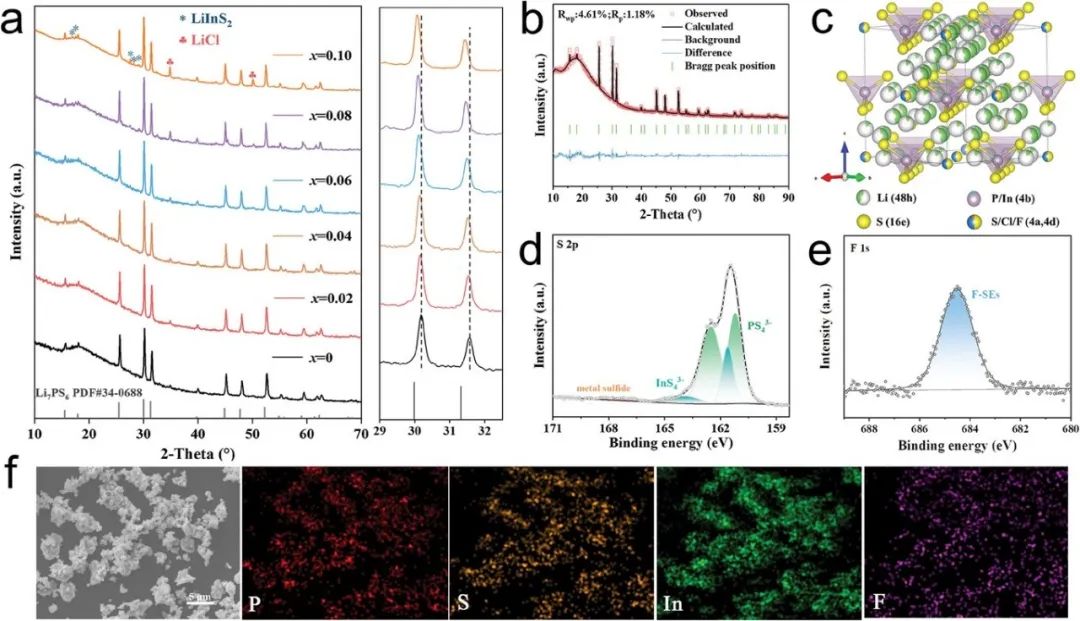
通過傳統的固相燒結方法合成了In和F共摻雜的鋰硫化物固態電解質Li5.7+2xP1-xInxS4.7Cl1.3-3xF3x(x = 0.02, 0.04, 0.06, 0.08, 和 0.1) 系列。通過粉末X射線衍射(XRD)圖譜驗證了摻雜濃度對LPSC結構的影響。不同比例摻雜的Li5.7+2xP1-xInxS4.7Cl1.3-3xF3x電解質均展示出類似的立方鋰-硫銀鍺礦型晶體結構(Li7PS6,PDF#34-0688),與參考的Li7PS6 (PDF#34-0688) 非常吻合。在晶體結構中,較小離子半徑F?(136 pm)對Cl?(181 pm)的取代將縮小電解質的晶胞體積,而較大的In3+(81 pm)對P5+(38 pm)的取代會抵消晶格體積的收縮,晶胞體積的變化是摻雜元素相互競爭的結果。隨著InF3摻雜量的增加,衍射峰持續向較低的衍射角移動,表明晶胞體積存在明顯的擴展現象。如圖S1所示,補充信息中,Li5.7+2xP1-xInxS4.7Cl1.3-3xF3x的晶格參數從x = 0時的9.825 ?穩步增加到x= 0.1時的9.856 ?,表明In3+摻雜在晶體晶格中起的作用比F-摻雜更為顯著。然而,由于In和F摻雜引起的溶解度限制,隨著摻雜量的增加(超過8%),包括LiCl、LiInS4、Li8P2S9、LiF等多種晶相逐漸出現。從熱力學的角度來看,使用DFT計算研究了在晶體結構不同位置摻雜In和F的可能性。補充信息圖S3展示了LPSC結構優化的不同摻雜模型。結果表明,摻雜的In原子傾向于占據P原子(4b位點),因為其摻雜穩定性更高。進一步,在優化的In摻雜LPSC結構中,F原子占據Cl 4a位點和Cl 4d位點的Ef分別為0.81和0.64 eV(補充信息圖S4b)。然而,F取代PS4基團和InS4基團中S位點所需的能量分別為-1.56和-1.24eV。因此引入F元素最有利于取代Cl 4a位點。Li5.7+2xP1-xInxS4.7Cl1.3-3xF3x電解質顆粒SEM和EDS圖譜表明,球磨后的電解質顆粒大小約為3-5 μm。XPS和EDS圖譜進一步證實了In和F元素成功摻雜進鋰硫化物固體電解質的晶體結構中。
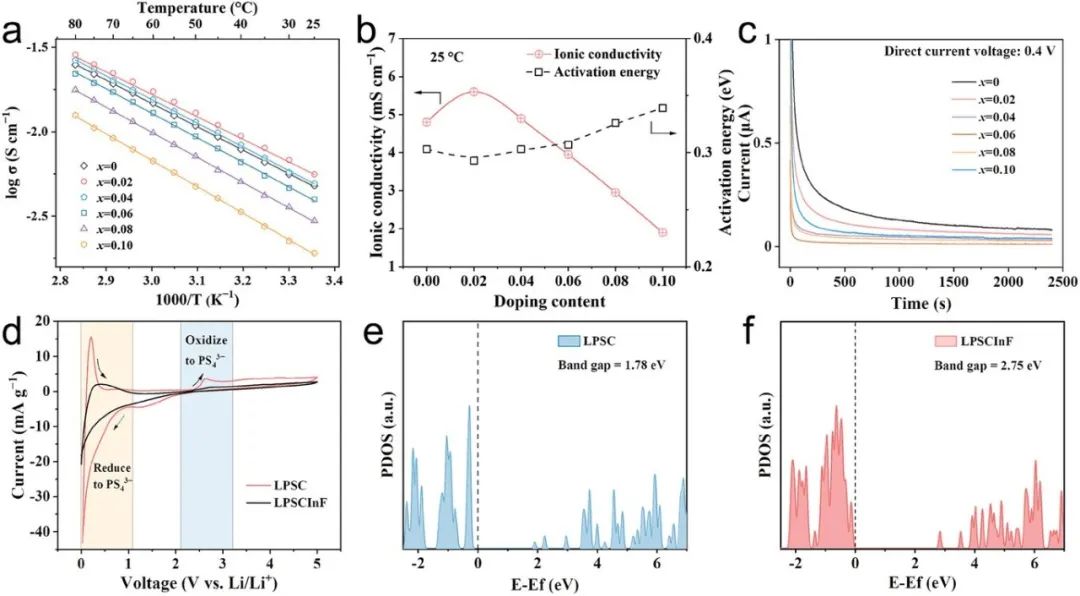
為了評估枝晶抑制能力,本文構建了對稱的Li/Li5.7+2xP1?xInxS4.7Cl1.3?3xF3x/Li電池,并進行了以0.1 mA cm?2的電流密度遞增的恒電流循環測試。CCD代表了ASSLBs能夠承受的最大電流密度,而不會因為枝晶生長導致電池失效,它決定了鋰金屬電池的功率密度。如圖3a所示,原始LPSC的CCD值為1.0 mA cm?2,這與文獻中先前報道的值相當。相比之下,隨著In和F濃度的增加,Li5.7+2xP1?xInxS4.7Cl1.3?3xF3x的CCD值也隨之增加,當x等于0.06時(LPSCInF,圖3b),達到最大值2.5 mA cm?2。
Li5.7+2xP1?xInxS4.7Cl1.3?3xF3x(x = 0.02, 0.04, 0.08, 0.1, 和 0.15)的電壓-時間曲線如圖S10所示,附錄信息中,所有不同摻雜量的硫化物SEs的CCD值都比原始LPSC高。值得注意的是,Li5.7+2xP1?xInxS4.7Cl1.3?3xF3x SEs的CCD值與電子導電性呈現相反的趨勢(圖S11,附錄信息)。
本文通過組裝鋰對稱電池進一步評估了Li5.7+2xP1?xInxS4.7Cl1.3?3xF3x固體電解質(SEs)的長期鋰鍍層/剝離循環性能。如圖S12所示,在支持信息中,Li/LPSC/Li對稱電池在電流密度為0.5 mA cm?2和截止容量為0.5mAh cm?2的條件下循環75小時后,顯示出不可逆的電壓下降,表明電池短路。與此形成鮮明對比的是,Li/Li5.7+2xP1?xInxS4.7Cl1.3?3xF3x (x = 0.02)/Li對稱電池在超過750小時的時間里沒有短路跡象(圖S13)。然而,在鋰鍍層/剝離過程中形成的界面相會導致極化電壓從23 mV逐漸增加到101 mV。在相同條件下,Li/LPSCInF (x = 0.06)/Li對稱電池可以在22 mV的平坦過電位下穩定運行超過2000小時,這展示了其抑制鋰枝晶生長的最佳能力(圖3d)。更值得注意的是,即使在高電流密度1 mA cm?2和截止容量1 mAhcm?2的條件下,Li/LPSCInF/Li對稱電池也能穩定運行超過1000小時(圖S14)。考慮到鋰金屬的電化學穩定性,通過In和F的共取代制備的硫化物SEs與報道中的最佳性能相當(圖3c;S2)。
為了理解LPSCInF抑制鋰枝晶生長的機制,進行了時間分辨的電化學阻抗譜(EIS)、掃描電子顯微鏡(SEM)、X射線光電子能譜(XPS)和密度泛函理論(DFT)計算。時間分辨的EIS測量也用于評估SEs與鋰金屬的界面穩定性(圖S15)。在最初的24小時內,Li/LPSCInF/Li電池的界面阻抗持續增加,并在循環72小時后穩定在約175 Ω,表明形成了界面相。相比之下,在48小時的儲存后,Li/LPSC/Li電池的阻抗從163 Ω繼續增長到312 Ω,表明LPSC/Li界面持續發生副反應。進一步,通過SEM測量來識別在0.5 mA cm?2/0.5mAh cm?2條件下對稱電池中鋰金屬的原始形態和循環50小時后的鋰金屬形態。在圖S16b中,支持信息中,Li/LPSCInF/Li對稱電池上沒有形成可見的鋰枝晶或裂紋,表明在Li/SEs界面上的鋰鍍層/剝離均勻。與此形成鮮明對比的是,由于界面接觸不均勻和Li+的不均勻沉積,可以在LPSC循環后的鋰金屬表面觀察到絨毛狀的鋰枝晶(圖S16c)。
在LPSCInF表面收集的XPS光譜是在與鋰金屬循環50小時后獲得的。Li 1s、P 2p、S 2p、Cl 2p、In 3d和F 1s光譜的總體結果在圖3e,f和圖S17中展示;支持信息中。如圖4e所示,通過F 1s光譜中684.8eV處觀察到的特征峰驗證了LiF的存在。Li 1s光譜中54.9 eV處的顯著信號也表明形成了LiF(圖S17a)。在支持信息中的圖S17b,c中,LPSCInF在與鋰金屬接觸時分解成Li2S和Li3P。LiCl、Li2S、Li3P和LiF的PDOS圖在支持信息中的圖S18中展示。在各種SEI成分中,LiF因其7.4 eV的寬帶隙而被認定為電子絕緣體。這些具有大帶隙的副產品(Li2S、LiF和LiCl)有效地阻斷了電子路徑并防止進一步分解,類似于傳統鋰離子電池中的固態電解質界面(SEI)。
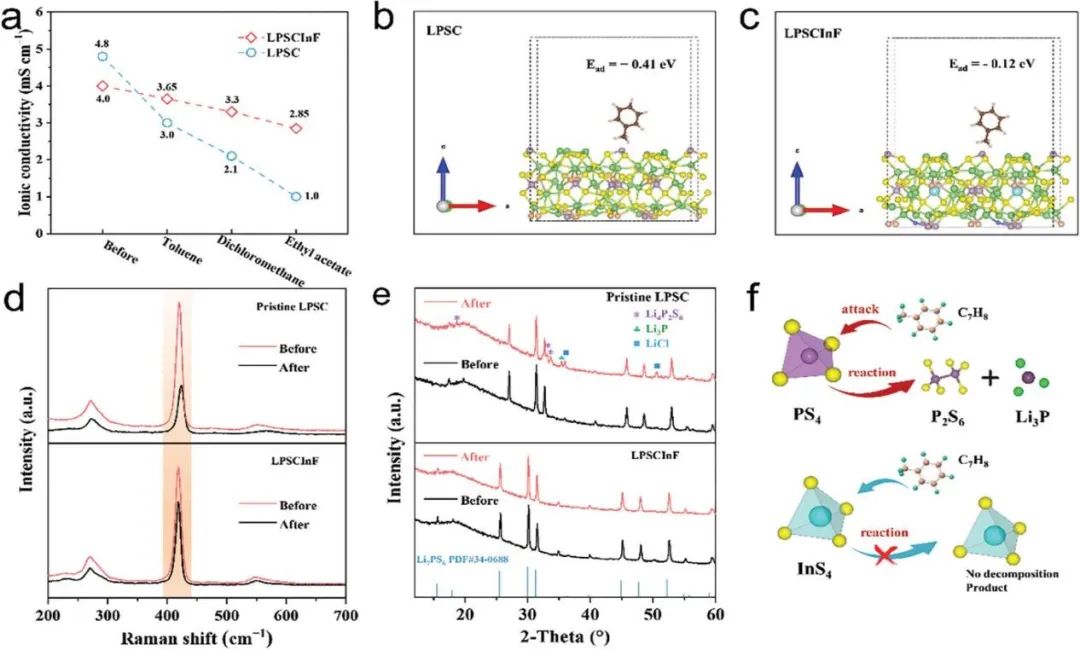
在表面,經過50小時的循環后,可以檢測到In3+、In0(450.7和443.0 eV)和Li-In合金(448.1和441.3 eV)三種化學狀態(圖3f)。Li-In合金相的體積化學擴散系數(D)約為10?8到10?6 cm2 s?1,這遠高于塊狀鋰金屬中的鋰(5.69 × 10?11 cm2 s?1)。在LPSCInF/Li界面生成的親鋰性Li-In合金有助于促進快速的傳輸動力學并調節鋰的均勻成核。根據以往的研究,Li(100)表面是最穩定的,因此用于后續的計算。我們計算了Li-In合金的表面能,Li-In合金(100)、(110)和(111)表面的表面能分別為0.50、0.40和0.51 eV ??2(圖S19a)。因此,Li-In合金(110)表面是最穩定的表面,被選用于鋰擴散計算。圖3g,h顯示了Li (100)表面和Li-In合金(110)表面的鋰擴散勢壘。計算的詳細信息在支持信息的實驗部分給出。Li-In合金中鋰原子的遷移勢壘沿(110)方向為0.17 eV,低于純鋰金屬(100)表面的0.38 eV勢壘。在支持信息的圖S19b中,Li-In合金(100)的遷移勢壘為0.23 eV,這略高于(110)方向。Li-In合金相較低的擴散能量勢壘有利于SEI層上Li+的擴散,進一步有助于鋰離子通量的均勻化。原位形成的電子阻擋LiF可以鈍化硫化物SEs與鋰金屬之間的電子傳輸。總體而言,使用In和F共摻雜策略制備的硫化物電解質對于實現鋰金屬陽極到高能量密度ASSLBs(圖3h)顯示出巨大的潛力。
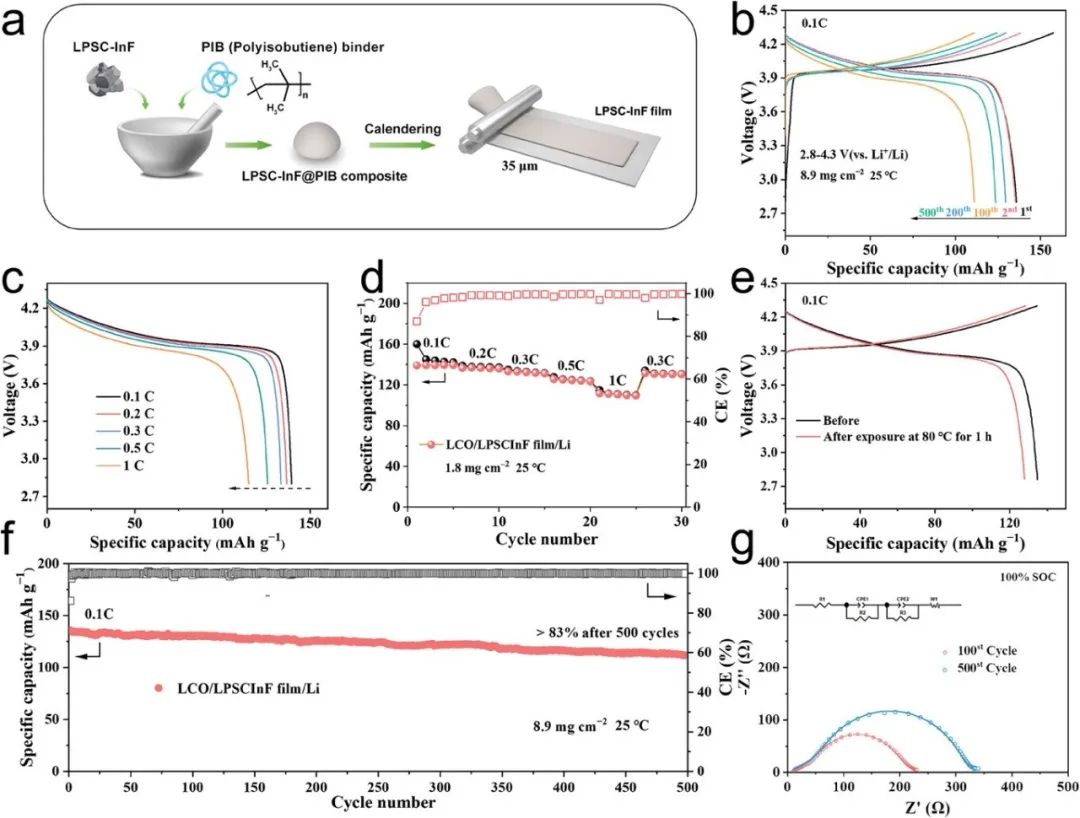
本文制備了由LPSCInF和PIB粘結劑構成的硫化物SE膜。PIB粘結劑的分子結構由具有小側鏈的飽和碳氫化合物組成,分子鏈之間存在較強的分子間吸引力,增強了分子之間的黏附力。
為了驗證LPSCInF-FIB復合固體電解質膜的電化學穩定性,使用LiCoO2(LCO)作為正極材料、Li金屬為負極及LPSCInF-FIB電解質膜組裝了全固態電池。組裝的全固態電池首圈放電比容量為135.7mAh g?1,初始庫侖效率為86.2%,經過500圈循環后容量保持率仍然高達83.2%。
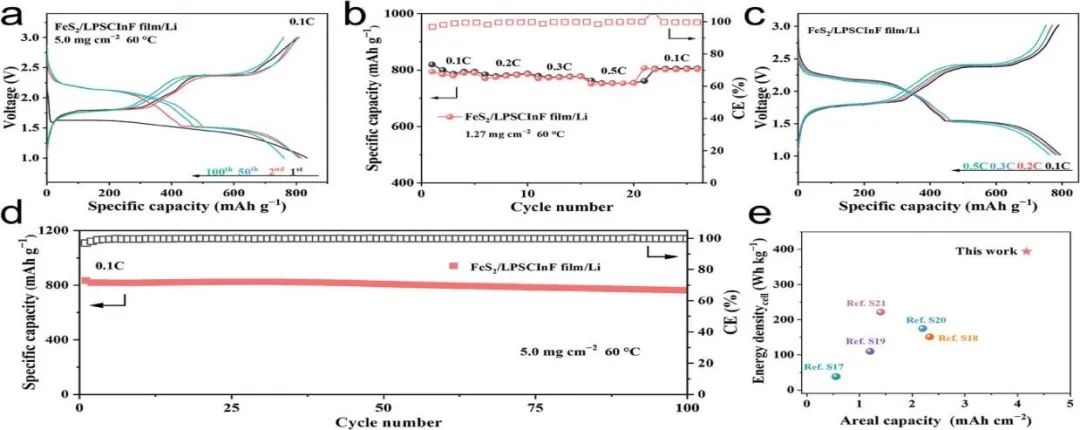
FeS2由于其超高的理論比容量894 mAh g?1、地球豐度和環境友好性,被選為構建高能量密度全固態鋰硫電池(ASSLBs)的陰極活性材料。在圖S33中,FeS2展示出單晶微結構,顆粒大小約為10微米。組裝的ASSLBs在1.0-3.0V(相對于Li+/Li)的電壓范圍內進行測試,電流為0.1 C和60 C。如圖6a所示,在第一個循環中,以FeS2為陰極的ASSLBs提供了834.1mAh g?1的放電比容量和96.9%的首次庫侖效率(ICE),活性材料負載為5.0 mg cm?2,這接近FeS2的理論比容量。在第二個放電曲線中,基于FeS2的ASSLBs顯示出兩個明顯的平臺,分別位于2.2 V和1.5 V,對應于中間形成的反應:FeS2 + 2Li+ + 2e? = FeS + Li2S,以及FeS + 2Li+ + 2e? = Fe0 + Li2S。然而,在50個循環后,放電電壓平臺向更高的電壓平臺移動,可能是由于金屬鐵單體的形成增加了復合正極的電子導電性。圖6b展示了FeS2/LPSCInF膜/Li電池在活性材料負載為1.27 mg cm?2時的倍率性能。通過逐漸將電流率從0.1增加到0.2、0.3和0.5 C,電池顯示出高比放電容量分別為819.7、785.2、779.7和763.0mAh g?1。不同電流率下的相應電壓曲線如圖6c所示。在0.5 C的高電流率下,與0.1 C相比,高容量保持率為93.1%,表明構建的ASSLBs具有優異的動力學性能。此外,圖6d展示了電池在0.1 C和60 C下的長期循環性能,經過100個循環后,放電容量保持率為91.3%。電池的相應電化學阻抗譜(EIS)結果如圖S34所示,其中電池在2個循環和100個循環時的阻抗分別為92.9 Ω和216.6Ω。這些結果表明,制備的LPSCInF SE膜具有優異的電化學穩定性,能夠與高體積變形的硫基活性材料兼容。基于電池參數(表S3,支持信息),采用LPSCInF膜的FeS2基ASSLBs顯示出高達410 Wh kg?1的高總質量能量密度,這在其他已報道的結果中處于高水平(圖6e;表S4)。
總結與展望
該研究成功制備了一系列摻雜了銦(In)和氟(F)的Li5.7+2xP1-xInxS4.7Cl1.3-3xF3x硫化物固態電解質(SEs),其離子導電性得到提高,對鋰金屬的電化學穩定性極佳。在硫化物基質中的最佳摻雜含量(x = 0.02)表現出最高的離子導電性,達到25°C時5.6毫西門子每厘米(mS cm?1)。在Li5.7PS4.7Cl1.3中,適度的銦和氟濃度(Li5.82P0.94In0.06S4.7Cl1.12F0.18,x = 0.06)使得鋰對稱電池在25°C時實現了超高的CCD值2.5毫安每平方厘米(mA cm?2)。即使在電流密度為1毫安每平方厘米和容量為1毫安時每平方厘米的情況下,鋰/LPSCInF/鋰對稱電池仍然可以穩定循環超過1000小時。通過漿料涂布和熱壓工藝制備了一種由摻雜硫化物SEs和PIB粘合劑組成的柔性硫化物復合薄膜(35微米)。組裝的LiCoO2/LPSCInF薄膜/Li電池在0.1 C下循環500次后,循環壽命長,為83.2%,并且具有優異的倍率性能。與FeS2陰極、鋰金屬陽極和薄SEs薄膜組裝的ASSLBs實現了410瓦時每千克(Wh kg?1)的能量密度和良好的循環性能(100個循環后》3.8毫安時每平方厘米)。這些結果展示了高性能硫化物SEs在制備用于快速充電和高能量密度ASSLBs的超薄硫化物SE薄膜中的重要性。
文獻鏈接Dabing Li, XinyuLiu, Yang Li, Xiaoxue Zhao, Meng Wu, Xiang Qi, Lei Gao, Li-Zhen Fan
AEM First published: 10 October 2024
原文鏈接:https://doi.org/10.1002/aenm.202402929
-
電解質
+關注
關注
6文章
821瀏覽量
20697 -
電池
+關注
關注
84文章
11044瀏覽量
134551
原文標題:北京科技大學范麗珍教授團隊In和F共摻雜LPSCl制備固體電解質
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
電子科技大學OpenHarmony技術俱樂部正式揭牌成立

南方科技大學: 研究基于聚電解質的無線和無漂移離電型傳感器,用于正畸傳感

清華大學:自由空間對硫化物固態電解質表面及內部裂紋處鋰沉積行為的影響
大學計劃 | 同星智能贊助電子科技大學 Fury 電動方程式賽車隊

馬里蘭大學王春生教授團隊最新研究成果:探索水系鋅電池的電解質設計

清華深研院劉思捷/港科大Kristiaan Neyts最新AEM封面文章:硫化物復合固態電解質

陳軍院士團隊最新Angew,聚合物電解質新突破

南方科技大學OpenHarmony技術俱樂部正式揭牌成立

Li3MX6全固態鋰離子電池固體電解質材料
華中科技大學:通過自組裝單層加強石墨烯器件的熱管理
NVIDIA CEO黃仁勛對話香港科技大學畢業生
胡先羅教授在AEM發表研究:探索寬溫電解質設計新路徑

固態電池中復合鋰陽極上固體電解質界面的調控






 工商網監
工商網監

評論