 機器學習模型可以幫助化學家更快地制造出具有更高功效的分子
機器學習模型可以幫助化學家更快地制造出具有更高功效的分子
設計新的藥物分子需要手工進行,耗時且容易出錯。但是麻省理工學院的研究人員已經朝著完全自動化的設計過程向前邁出了一步,這將大大加快設計過程,并獲得更好的結果。機器學習模型可以幫助化學家更快地制造出具有更高功效的分子。
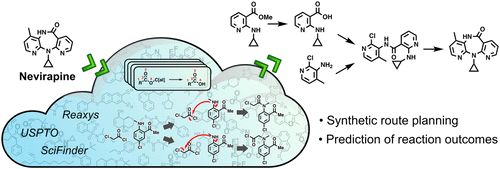
藥物發現依賴于先導化合物的優化。根據先導化合物與特定的生物目標的相互作用,增減官能團,手動調整分子結構來獲得新的分子。每個修飾步驟都要耗費大量時間,并且往往得不到期望中的分子。
麻省理工學院計算機科學與人工智能實驗室( CSAIL )和電子工程與計算機科學系( EECS )的研究人員開發了一種模型用于更有效的選擇先導分子。該模型輸入分子結構數據,創建分子圖片,詳細展示分子結構,節點代表原子,邊線代表化學鍵。這些圖又被分解成更小的有效官能團簇,成為“構件”,用于實現更精確的分子重構和修飾。

“設計這個模型的目的,是想用自動迭代取代低效的人工分子改造過程,并確保設計出有效的分子,”CSAIL博士生,本文的主要作者Wengong Jin說。該模型在7月份舉行的2018國際機器學習會議上進行了展示。論文的共同作者還包括CSAIL和EECS的Delta電子系教授Regina Barzilay和電氣工程和計算機科學,數據、系統和社會研究所的Tommi S. Jaakkola教授。
這項研究是麻省理工學院與八家制藥公司于五月宣布的藥物發現與合成機器學習聯盟的其中一部分工作。該聯盟將先導物的優化確定為藥物發現的一個關鍵挑戰。
Barzilay說:“ 目前來說,需要許多熟練的化學家的大量工作才能實現先導物的優化,而這正是我們想要改進的地方。下一步的計劃,是讓該技術從學術界走向真正的藥物設計實踐,并證明它可以幫助人類化學家完成該項工作,而這將是一個挑戰。"
Jaakkola說:“程序自動化也為機器學習帶來了挑戰。通過學習關聯、修改和生成分子圖推動新的技術思想和方法的產生。"
生成分子圖
近年來,分子設計自動化的系統層出不窮,但問題是產生的分子是否有效。Jin說,這些系統通常會產生符合化學規則但實質無效的分子,并且生成的分子不具備最佳性質。這實際上使完全自動化設計分子變得行不通。
這些系統運行在分子的線性符號上,稱為“簡化分子-輸入線-進入系統”(simplified molecular-input line-entry systems, SMILES),其中長串的字母、數字和符號代表可由計算機軟件解釋的單個原子或化學鍵。當系統修改一個前導分子時,它會一個符號一個符號地擴展它的字符串表示,一個原子一個原子,一個鍵一個鍵,直到生成一個最終的具有更高的期望屬性的SMILES字符串。最后,系統可能會生成一個最終的SMILES字符串,雖然在SMILES語法下似乎是有效的,但實質上是無效的分子。
研究人員通過建立一個直接在分子圖上運行的模型來解決這個問題,和SMILES字符串不同,該方法可以更有效和準確的修改分子結構。
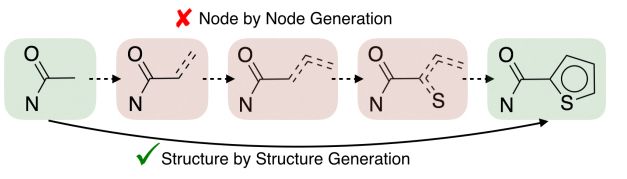
模型的核心是一個定制的可變自動編碼器——一個神經網絡,它將輸入分子“編碼”成向量,這個向量是分子結構數據的存儲空間,然后將該向量“解碼”成與輸入分子匹配的圖形。
在編碼階段,模型將每個分子圖分解成簇或“子圖”,每個簇或子圖代表一個特定的構建元件。這樣的簇是由一個常規的機器學習“樹分解”來自動構建的,其中復雜的圖被映射成一個簇的樹結構,提供了原始的分子圖支架。
支架樹結構和分子圖結構都被編碼到它們自己的向量中,其中分子根據相似性被分在一組。使得尋找和修飾分子變得更容易。
在解碼階段,該模型以“由粗到細”的方式重建分子圖——逐漸增加低分辨率圖像的分辨率以創建更精細的版本。它首先生成樹形結構的支架,然后將關聯的簇(樹中的節點)組裝在一起形成一個連貫的分子圖。確保重建的分子圖是原始結構的精確復制。

該模型可以基于期望的性質修改先導分子。通過預測算法,用期望性質的效力值對每個分子進行評分。例如,在這篇論文中,研究人員尋找具有兩種性質的分子——高溶解度和可合成性。
對于給定的期望性質,該模型通過使用預測算法來優化先導分子,從而通過編輯分子的官能團來修改向量,從而修改結構,以獲得更高的效力分數。重復這個步驟經過多次迭代,直到找到最高的預測效能分數。然后,模型通過編譯所有相應的簇,最終從更新的向量中解碼出一個修改了結構的新分子。
正確且有效
研究人員在ZINK數據庫中的25萬個分子圖上訓練了他們的模型,ZINK數據庫是一個可供公眾使用的三維分子結構集合。他們對模型進行了任務測試,以產生有效分子并找到最佳前導分子,設計出具有增強能力的新型分子。
在第一次測試中,研究人員的模型從樣本分布中產生了100 %的化學有效分子,而SMILES模型從相同分布中只產生43 %的有效分子。
第二項測試包括兩項任務。首先,該模型搜索整個分子集合,以找到所需性質的最佳先導分子——溶解性和可合成性。在這項任務中,該模型發現了一種先導分子,其效力性能比傳統系統高出30 %。第二項任務是修飾800個分子來獲得更高的分子效力性能,但結構類似于先導分子。在此過程中,該模型創造了與先導分子結構非常相似的新分子,且分子平均效力提高了80 %以上。
除了溶解度以外,研究人員接下來的目標是測試模型的更多性質,這些性質與醫療關系更密切。但是這也需要更多的數據。制藥公司對能夠對抗生物靶點的分子特性更感興趣,但他們掌握的這類數據較少。研究人員面對的挑戰是開發一種能夠在有限的訓練數據下工作的模型。
未參與此項研究的Amgen醫藥公司的醫藥化學主管Angel Guzman-Perez說:“論文中描述的算法朝著模仿先導分子優化設計的目標邁出了重要的一步,而這個工作目前是醫藥化學家在做的。由于這種計算方法在向量空間中進行分子性能優化,它有可能設計出完全不同和新穎的化學結構,這是藥物化學家在化學結構空間中考慮問題時所無法企及的。因此,這種算法可以補充和提升藥物化學家的工作。”
-
人工智能
+關注
關注
1791文章
47350瀏覽量
238754 -
計算機科學
+關注
關注
1文章
144瀏覽量
11370 -
機器學習
+關注
關注
66文章
8422瀏覽量
132714
原文標題:新藥研發的加速器:MIT研究人員開發機器學習方法,實現分子設計自動化
文章出處:【微信號:thejiangmen,微信公眾號:將門創投】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
請問ARM是怎么制造出來的?
臺灣科學家以水熱法制造出白光LED
血細胞的產生與美國科學家成功制造出具有造血干細胞功能的細胞
科學家制造出鴿子機器人 一種能模仿鳥類飛行方式的飛行器
日本制造出能感知疼痛的機器人 人與物體之間的界限更小了
日本制造出了一款能夠感知疼痛的智能機器人
韓國科學家制造出電極,或能延長鋰空氣電池的壽命
如何制造出具有更高采樣率的示波器?
科學家意外發明新材料,可制造更快的芯片






 工商網監
工商網監
評論