 電子發(fā)燒友App
電子發(fā)燒友App


?
研究背景
水系鋅離子電池由于其高度的安全性和環(huán)境友好性,已成為部署大規(guī)模儲(chǔ)能設(shè)備的強(qiáng)有力的候選者。鋅負(fù)極因其高容量、高存儲(chǔ)容量和低還原潛力而備受關(guān)注。近年來,鋅負(fù)極的改性研究取得了很大進(jìn)展,包括電解液添加劑、電極結(jié)構(gòu)設(shè)計(jì)和人工電極表面改性。無機(jī)涂層表面改性是目前最有前途的實(shí)用策略之一,受到了廣泛的關(guān)注。然而,無機(jī)涂層的剛性和低離子電導(dǎo)率限制了鋅負(fù)極在大電流和大容量下的循環(huán)。
?
成果簡(jiǎn)介
鑒于此,哈爾濱工業(yè)大學(xué)的張乃慶教授(通訊作者)團(tuán)隊(duì)在Zn負(fù)極上構(gòu)建了具有類SEI結(jié)構(gòu)的PVA@SR-ZnMoO4多功能涂層(SR指SO42-受體)。PVA@SR外層可以使涂層具有一定的柔性,同時(shí)增強(qiáng)了Zn2+的流動(dòng)性。且ZnMoO4內(nèi)層能有效抑制枝晶生長(zhǎng)和副反應(yīng)。相關(guān)成果以“Multifunctional SEI-like bilayer structure coating stabilizing Zn anode at large current and capacity”為題發(fā)表在Energy & Environmental Science上。
?
研究亮點(diǎn)
1、類SEI結(jié)構(gòu)的設(shè)計(jì)可以有效提升修飾層的機(jī)械穩(wěn)定性,有效適應(yīng)高面容量循環(huán)條件下鋅負(fù)極的體積變化;
2、ZnMoO4與PVA凝膠之間建立了快速Zn2+遷移通道,改善了ZnMoO4的緩慢離子傳導(dǎo)。
?
圖文介紹

方案1 裸鋅和PVA@SR-ZnMoO4?SEI型結(jié)構(gòu)涂層改性鋅的循環(huán)示意圖。@RSC
金屬鋅負(fù)極的不穩(wěn)定性限制了水系鋅離子電池的進(jìn)一步發(fā)展。如方案1所示,動(dòng)力學(xué)和熱力學(xué)上都利于枝晶的生長(zhǎng),成為影響電池壽命的主要因素。在之前的報(bào)道中,ZnMoO4已被證明,能有效抑制枝晶生長(zhǎng)和氫氣的析出,使其成為無機(jī)組分的理想選擇。考慮到PVA凝膠具有良好的Zn2+轉(zhuǎn)運(yùn)性和包覆穩(wěn)定性,選擇PVA凝膠作為有機(jī)組分。此外,SR被添加到SO42-加速反離子Zn2+的遷移。因此,所構(gòu)建的類SEI結(jié)構(gòu)涂層有望在大電流和大容量下穩(wěn)定Zn負(fù)極。
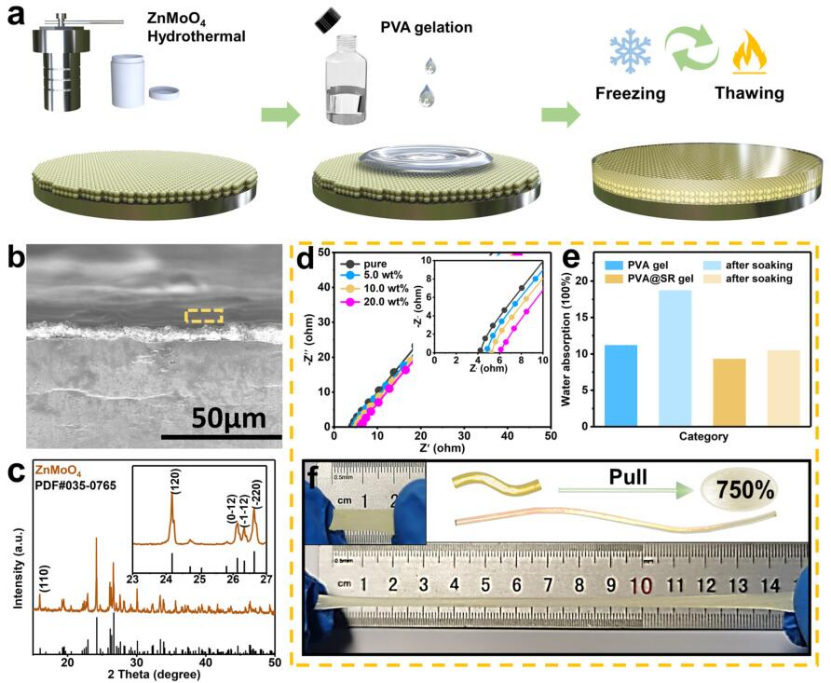
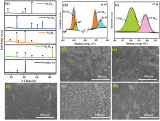
圖1 (a)類SEI結(jié)構(gòu)修飾層改性鋅負(fù)極的制備過程示意圖;(b)類SEI結(jié)構(gòu)修飾層的SEM照片;(c)ZnMoO4的XRD譜圖;(d)不同SR添加量的PVA凝膠的EIS譜;(e)純PVA和SR改性PVA的吸水性測(cè)試;(f)PVA@SR凝膠的延展性。@RSC
作者采用兩次刮刀涂布獲得了類SEI涂層,過程中PVA凝膠需要通過三個(gè)凍融循環(huán),進(jìn)一步成型(圖1a)。如圖1c所示,ZnMoO4的XRD譜圖與標(biāo)準(zhǔn)卡片匹配,鍍層厚度約為8 μm(從圖1b得知)。如圖1d所示,PVA凝膠的離子電導(dǎo)率隨著SR的加入逐漸增加,在10 wt%時(shí)達(dá)到峰值(1.419*10-2?S cm-1),甚至高于3.0 M ZnSO4水基電解質(zhì)(1.182*10-2?S cm-1)。如圖1e所示,純PVA凝膠和改性PVA凝膠在初始狀態(tài)下的含水量基本相當(dāng)。純PVA凝膠在去離子水中浸泡7天后,其含水量明顯增加,由此產(chǎn)生的溶脹會(huì)嚴(yán)重影響涂層穩(wěn)定性。相比之下,改性PVA凝膠幾乎沒有發(fā)生溶脹。SR加入后,PVA凝膠的延展性也有所提高,從500%增加到750%(圖1f),進(jìn)一步證明了改性PVA凝膠可以作為有機(jī)組分來構(gòu)造類SEI涂層。
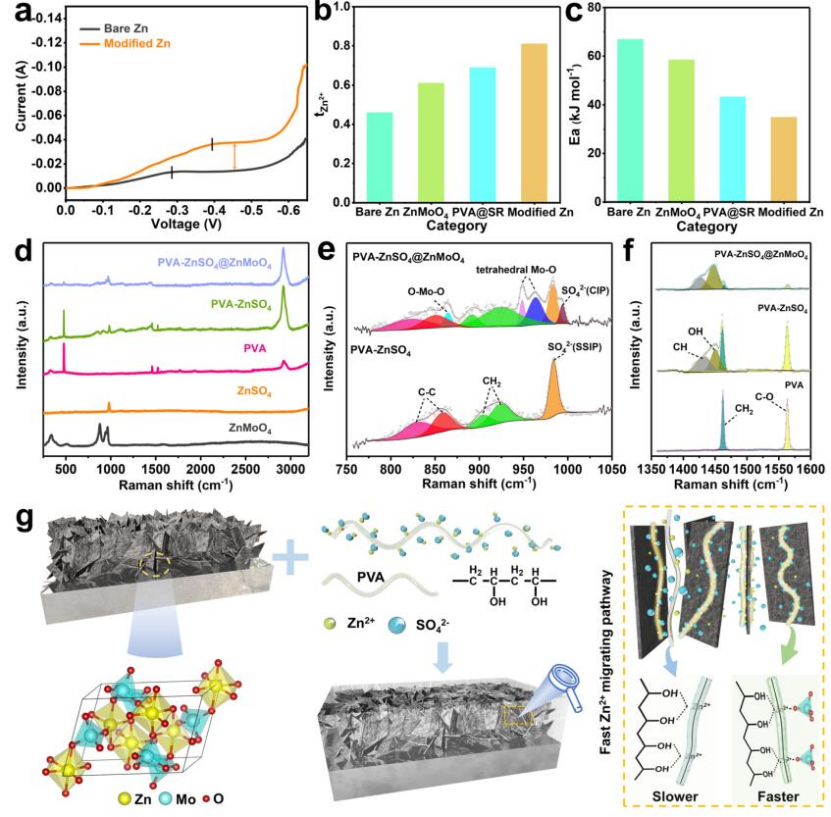
圖2 (a)?1 mV s-1掃速下,在1M ZnSO4電解質(zhì)中組裝的對(duì)稱電池;(b)Zn2+遷移數(shù)和(c)不同修飾的鋅負(fù)極的活化能;(d-f)拉曼光譜;(g)ZnMoO4/PVA界面遷移路徑中鋅離子遷移示意圖。@RSC
為了證明凝膠有機(jī)組分的快速離子輸送能力,組裝了對(duì)稱電池進(jìn)行驗(yàn)證。如圖2a所示,在1 mVs-1時(shí),裸鋅在-0.286 V處出現(xiàn)明顯的擴(kuò)散限制電流平臺(tái),且電流僅為-0.013 A。而改性的鋅負(fù)極具有更高的電流平臺(tái)(-0.038 A)。這表明類SEI結(jié)構(gòu)涂層會(huì)具有凝膠組分的高離子傳輸能力。作者計(jì)算了Zn2+遷移數(shù)來驗(yàn)證ZnMoO4無機(jī)組分對(duì)Zn2+遷移的貢獻(xiàn)。純鋅負(fù)極具有緩慢的Zn2+遷移能力(tzn2+為0.46),而類SEI涂層改性鋅負(fù)極離子遷移率高達(dá)0.81(圖2b)。改性鋅的活化能(Ea)僅為34.9 kJ mol-1,遠(yuǎn)低于裸鋅的(66.9 kJ mol-1),表現(xiàn)出較高的電化學(xué)反應(yīng)活性(圖2c)。
為了探究Zn2+在類SEI結(jié)構(gòu)涂層中的快速遷移機(jī)理,采用拉曼光譜研究了無機(jī)組分與有機(jī)組分之間的內(nèi)在聯(lián)系。在ZnSO4引入PVA后,出現(xiàn)了C-H(1432 cm-1)和O-H(1450 cm-1)信號(hào)峰,表明Zn2+在PVA中以-OH基團(tuán)為中心的遷移形式(圖2d)。進(jìn)一步引入ZnMoO4后,PVA的C-O鍵彎曲振動(dòng)峰(478 cm-1)和CH2剪切信號(hào)峰(1462 cm-1)(圖2f)幾乎消失,證明了PVA與ZnMoO4之間存在相互作用。ZnMoO4中引入PVA后,PVA的拉曼信號(hào)峰在面積和位置上都發(fā)生了變化,這些變化是由PVA的-OH基團(tuán)與ZnMoO4的Mo-O四面體相互作用引起的。ZnMoO4與PVA之間的相互作用改變了它們的界面環(huán)境,在-OH和Mo-O之間構(gòu)建了Zn2+快速遷移路徑,加速了Zn2+的遷移并促進(jìn)了Zn2+的脫溶劑化(圖2g)。
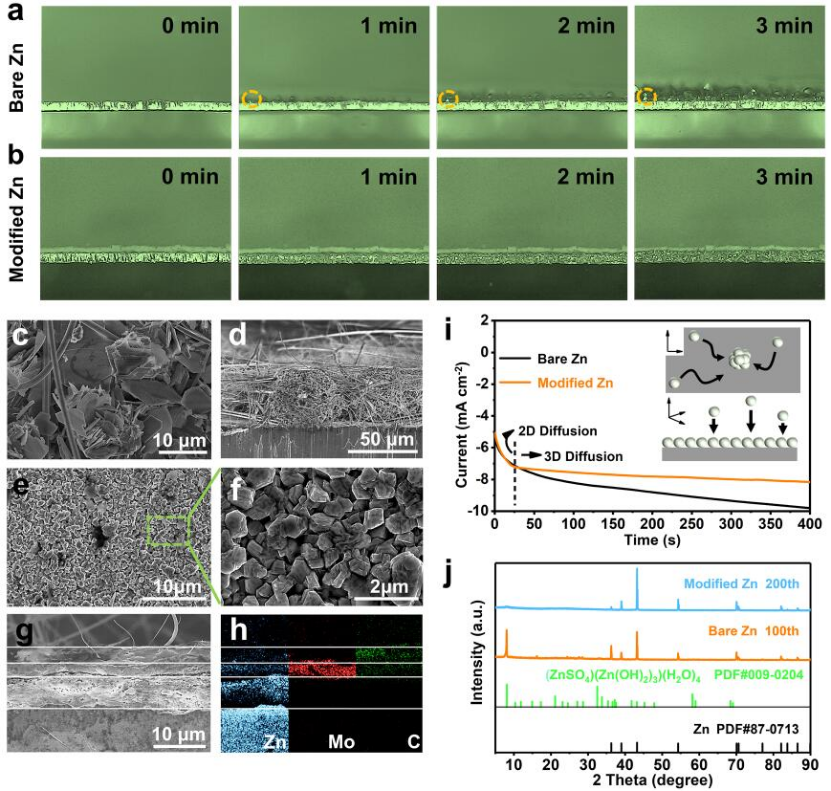
圖3 在50 mA cm-2下,(a)純Zn和(b)改性Zn的原位光學(xué)顯微鏡;(c,d)0.25 mA cm-2和5.0 mA h cm-2的純Zn負(fù)極沉積SEM圖像;(e-g)0.25 mA cm-2和5.0 mA h cm-2的改性Zn負(fù)極沉積SEM圖像和EDS圖譜(h);(i)計(jì)時(shí)安培法(CA)圖;(j)循環(huán)后的XRD圖(改性Zn負(fù)極)。@RSC
作者進(jìn)一步探究了類SEI結(jié)構(gòu)涂層對(duì)枝晶和副反應(yīng)的抑制作用。由原位光學(xué)顯微鏡實(shí)時(shí)監(jiān)測(cè)鋅沉積過程可知,在沉積的初始階段(1分鐘前),純Zn負(fù)極上出現(xiàn)了氣泡和尖銳突起(用虛線圓圈標(biāo)記)(圖3a)。隨著沉積的繼續(xù),出現(xiàn)了樹枝狀突起,且有氫氣析出。而類SEI結(jié)構(gòu)涂層修飾的Zn負(fù)極,即使在沉積時(shí)間達(dá)到8 min(沉積容量相當(dāng)于6.67 mA h cm-2)時(shí),鋅沉積層仍然保持相對(duì)平坦,沒有枝晶形成(圖3b)。
原位掃描電鏡圖像也顯示,沉積容量為0.25 mA h cm-2時(shí),裸鋅表面存在明顯的樹枝狀形態(tài)。而經(jīng)類SEI結(jié)構(gòu)涂層修飾的Zn負(fù)極表面在沉積容量為5 mA h cm-2時(shí)仍保持平坦(圖3e-h)。并且鋅的沉積形態(tài)由片狀變?yōu)閴K狀,降低了形成有害枝晶的可能性。此外,相對(duì)致密的沉積層抑制了氫的析出。
采用計(jì)時(shí)安培(CA)測(cè)試以了解裸Zn和改性Zn負(fù)極的成核行為。如圖3i所示,電流的連續(xù)變化表明,在裸鋅中發(fā)生了Zn2+2D擴(kuò)散,Zn2+沿負(fù)極表面遷移,并以較高的表面能積累,引發(fā)枝晶形成,導(dǎo)致粗糙的沉積形態(tài)。而ZnMoO4具有較高的鋅親和力,Zn2+直接原位還原為Zn0,無需選擇特殊位置。Zn2+在短暫的2D擴(kuò)散后轉(zhuǎn)向穩(wěn)定的3D擴(kuò)散,形成了致密的形核位點(diǎn)和光滑的沉積形態(tài)。裸鋅在100次循環(huán)后,表現(xiàn)出明顯的(ZnSO4)(Zn(OH)2)3(H2O)4副產(chǎn)物,而改性鋅即使在200次循環(huán)后也沒有產(chǎn)生副產(chǎn)物(圖3j)。
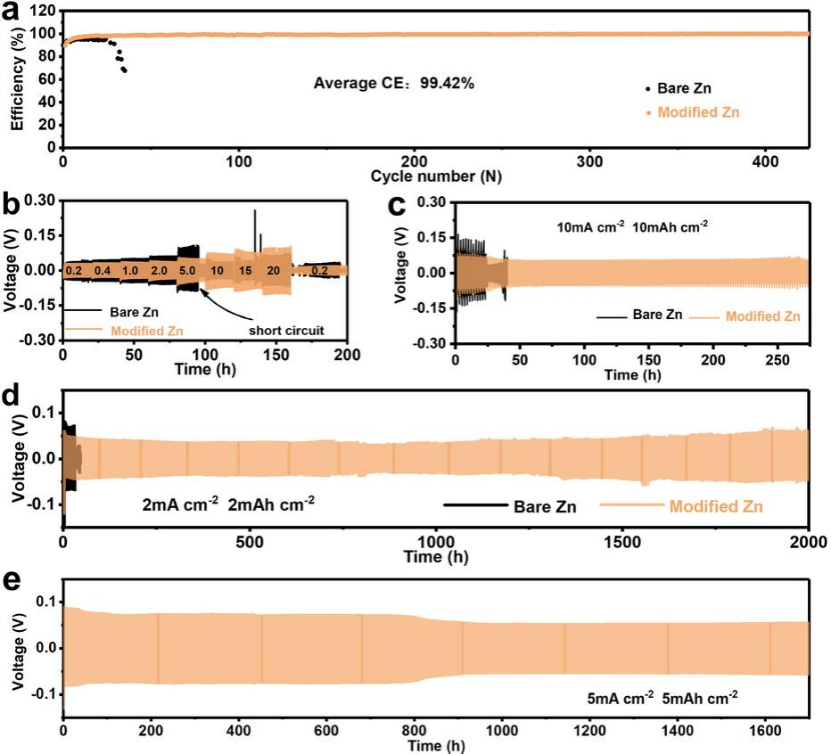
圖4 (a)在0.5 mA cm-2和0.5 mA h cm-2下,純鋅和改性鋅的庫(kù)倫效率;(b)對(duì)稱電池的倍率性能;(c)在10 mA cm-2和10 mA h cm-2下,鋅對(duì)稱電池的電壓分布。對(duì)稱電池的長(zhǎng)期恒流循環(huán)性能:(d)2?mA?cm-2,2 mAh cm-2、(e)5?mAcm-2和5?mAh cm-2。@ RSC
作者組裝了Zn/Ti不對(duì)稱電池,以研究電池的長(zhǎng)循環(huán)性能(圖4a)。裸鈦電池表現(xiàn)出較差的循環(huán)穩(wěn)定性,在0.5 mA cm-2的電流密度下50個(gè)周期內(nèi)庫(kù)倫效率(CE)顯著下降。而類SEI結(jié)構(gòu)涂層改性Ti可將CE提高到99.42%,且穩(wěn)定運(yùn)行500次。
圖4b展示了對(duì)稱電池的倍率性能,由于改性鋅電池加速了Zn2+的遷移,改性鋅電池的滯后電壓小于裸鋅電池。此外,隨著電流密度的增加,裸鋅電池在5.0 mA cm-2時(shí)迅速短路失效。而改性鋅在20 mA cm-2的高電流密度下,其電壓仍然保持相對(duì)穩(wěn)定。如圖4c所示,改性鋅電池在10mAcm-2和10mAh cm-2可穩(wěn)定循環(huán)275 h,而裸鋅電池在30?h后出現(xiàn)短路。
如圖4d所示,裸鋅電池的循環(huán)性能較差,在2?mA?cm-2和2?mAh cm-2時(shí)循環(huán)50?h就發(fā)生了短路。而改性的鋅負(fù)極表現(xiàn)出良好的循環(huán)穩(wěn)定性,可將循環(huán)時(shí)間延長(zhǎng)至2000 h,且電壓相對(duì)穩(wěn)定。改性后的鋅電池可以在5?mAcm-2和5?mAh cm-2時(shí)下循環(huán)1700 h,顯示出足夠的實(shí)際應(yīng)用潛力。
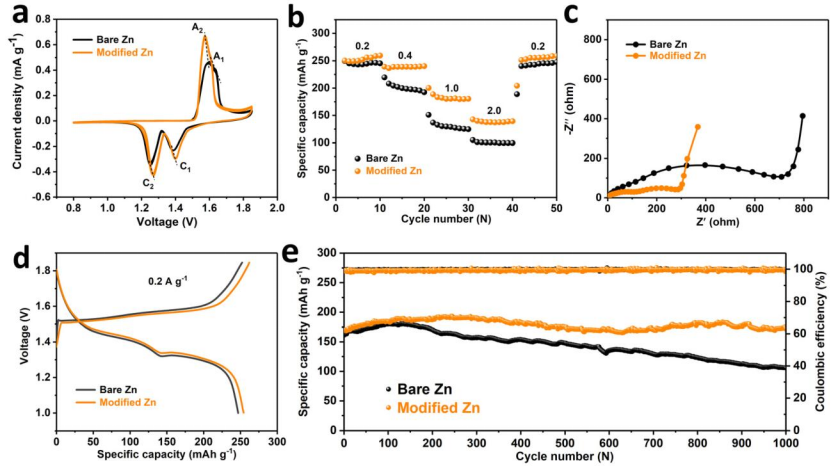
圖5?采用裸鋅和改性鋅負(fù)極Zn/α-MnO2電池的電化學(xué)性能:(a)0.1 mV s-1時(shí)的CV曲線;(b)倍率性能;(c)循環(huán)前的EIS曲線,(d)活化后0.2 A g-1時(shí)的充放電曲線。(e)1.0 A g-1時(shí)的長(zhǎng)循環(huán)性能。@ RSC
采用α-MnO2為正極材料組裝了全電池,并進(jìn)行相關(guān)的電化學(xué)測(cè)試。如圖5a所示,循環(huán)伏安曲線(CV)均出現(xiàn)了兩對(duì)氧化還原峰(A1, C1和A2, C2),分別表示H+和Zn2+在α-MnO2正極材料中的嵌入和脫出。與裸鋅電池相比,改性鋅負(fù)極電池具有更小的氧化還原峰電位間隙,從而表現(xiàn)出更小的電化學(xué)極化和更高的反應(yīng)活性。
圖5c中的電化學(xué)阻抗譜(EIS)也清楚地表明,改性鋅電池具有更快速的電荷轉(zhuǎn)移過程。改性鋅電池0.2 A g-1時(shí)的放電容量為255.21 mA h cm-2高于裸Zn負(fù)極電池(246.82 mA h cm-2)。改性鋅電池具有較高的反應(yīng)活性和較高的放電容量,具有較好的倍率性能。在不同電流密度下,改性鋅電池的放電容量均高于裸鋅電池,且容量波動(dòng)較小(圖5b)。改性鋅電池還表現(xiàn)出優(yōu)異的長(zhǎng)周期穩(wěn)定性,在1.0 A g-1下1000次循環(huán)后,保持141.7 mA h g-1的容量,而裸鋅電池的容量保持率僅為59.1%(圖5e)。
總結(jié)與展望
本工作構(gòu)造了具有類似SEI結(jié)構(gòu)的PVA@SR-ZnMoO4涂層可以有效地穩(wěn)定Zn負(fù)極。具有良好柔韌性的PVA@SR外層可以補(bǔ)償無機(jī)組分的剛性,提高大容量循環(huán)下的涂層穩(wěn)定性。無機(jī)ZnMoO4還能有效抑制枝晶和副反應(yīng)。ZnMoO4與PVA的相互作用可以建立Zn2+的快速遷移途徑,促進(jìn)Zn2+的脫溶。本工作為鋅負(fù)極表面改性提供了一種實(shí)用的設(shè)計(jì)策略,為實(shí)現(xiàn)穩(wěn)定快速的大容量循環(huán)提供了可能,拓寬了高離子導(dǎo)電性復(fù)合材料的設(shè)計(jì)思路。
文獻(xiàn)鏈接
Multifunctional SEI-like structure coating?stabilizing Zn anodes at a large current and?capacity.?(Energy Environ. Sci., 2022.?DOI:?10.1039/d2ee02931f.)
原文鏈接:
https://doi.org/10.1039/d2ee02931f
編輯:黃飛
?





















 工商網(wǎng)監(jiān)
工商網(wǎng)監(jiān)
評(píng)論