研究背景
近年來,鋅離子電池(ZIBs)迅速發展,成為高容量、高安全性及低成本電池的潛在替代品,但其仍然存在能量密度不足、庫侖效率差、鋅離子轉移/擴散速率緩慢及生長枝晶等缺點,因此,需要針對上述缺點進行改性。
電解質中陰離子對電池長期連續運行起著關鍵作用,對于鋅離子電池,它們經常與陽離子形成強耦合,導致溶劑化殼內靜電干擾。因此,陰離子隨機運動會對Zn2+的流暢和快速擴散/轉移造成嚴重阻礙,最終導致陽離子轉移數低和庫倫效率差。改善以上問題的方法主要有:
(1)固定陰離子的策略,該方法已應用于各種金屬離子電池的固態電解質,以實現合適的陽離子轉移數,也有助于緩解空間電荷區的陽離子消耗,可以抑制枝晶生長。
(2)可逆形成優先晶體取向的沉積層,是抑制枝晶生長的有效措施。具體而言,設計具有較低表面能和較高堆疊密度的Zn(002)晶面,可以成為有效抑制Zn負極表面發生的枝晶生長/副反應的解決方案。
盡管研究者在獲得取向Zn(002)結構方面已經付出了巨大努力,但大多數改性效果并不令人滿意,且對Zn(002)晶面優先生長的機制尚不明確,需要進一步推動開發更多晶面控制工程,同時明確內部調控機制。本工作通過將陰離子β-環糊精引入Zn(ClO4)2體系,提出了一種有趣的晶面取向及電解質工程設計方法。
成果簡介
近期,暨南大學麥文杰研究員和香港城市大學支春義教授在Angew上發表了題為“Anion-trap Engineering toward Remarkable Crystallographic?Reorientation and Efficient Cation Migration of Zn Ion Batteries”的文章。該工作受主客體相互作用化學的啟發,通過將陰離子捕集劑β-環糊精(β-CD)引入Zn(ClO4)2電解質中,將陰離子ClO4-限域于β-CD空腔內,以削弱Zn2+遷移壁壘,Zn2+遷移數顯著提高至0.878,且β-CD@ClO4配合物優先生長Zn(002),阻止了枝晶生長。上述協同作用顯著提高了鋅離子電池長期穩定性和電池容量,對開發新型鋅離子電池體系具有重要作用。
圖文導讀
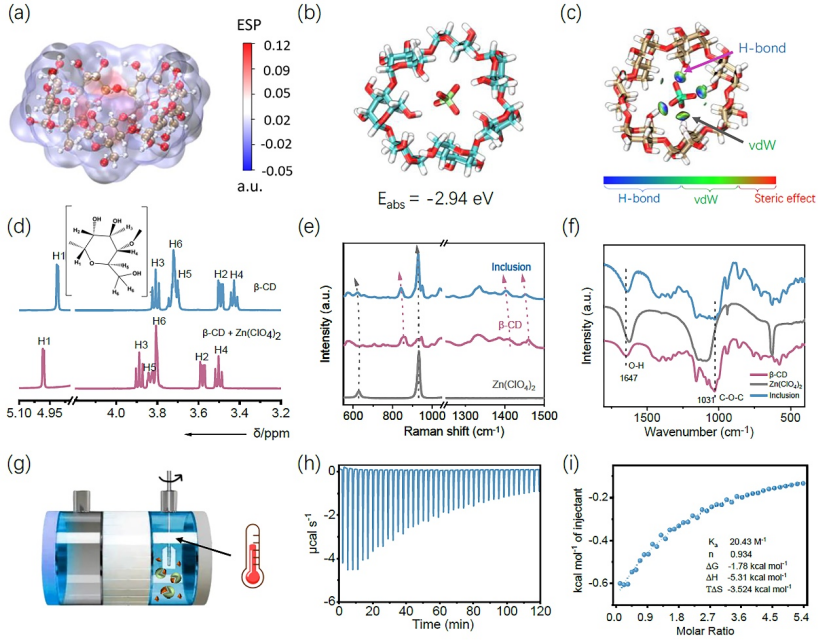
圖1. β-CD@ClO4的主客體相互作用的理論計算和表征。(a) β-CD的靜電勢圖;(b) β-CD@ClO4-的結構圖和結合能;(c) β-CD@ClO4-配合物IGM分析;(d) 游離β-CD及其配合物1H NMR譜圖;(e)游離β-CD、Zn(ClO4)2和β-CD@ClO4-配合物的拉曼光譜;(f)紅外光譜;(g) 微量熱滴定實驗裝置示意圖;(h) Zn(ClO4)2連續加入β-CD溶液的原始ITC數據;(i) 量熱曲線積分計算表觀反應熱。
β-CD是一種環狀寡聚體,由七個α-D-吡喃葡萄糖殘基連接,呈淺截錐狀,具有疏水內腔。根據獨立梯度模型(IGM)分析可知,β-CD與陰離子之間的弱相互作用以氫鍵為主,具有部分范德瓦耳斯力。為進一步驗證β-CD主體與ClO4-客體的結合行為,進行了核磁共振、拉曼和紅外光譜分析。結果表明,ClO4-進入其內腔并成功形成氫鍵,具有明顯主客體相互作用,且β-CD@ClO4-絡合物的形成對β-CD骨架結構沒有影響。
此外,通過等溫滴定量熱法(ITC)進一步研究β-CD和ClO4-之間相互作用行為,這是表征主客體相互作用熱力學性質最直接和定量的方法,分析量熱數據可知,β-CD和ClO4-結合是一個焓驅動過程(DH=-5.31 kcal mol-1)。
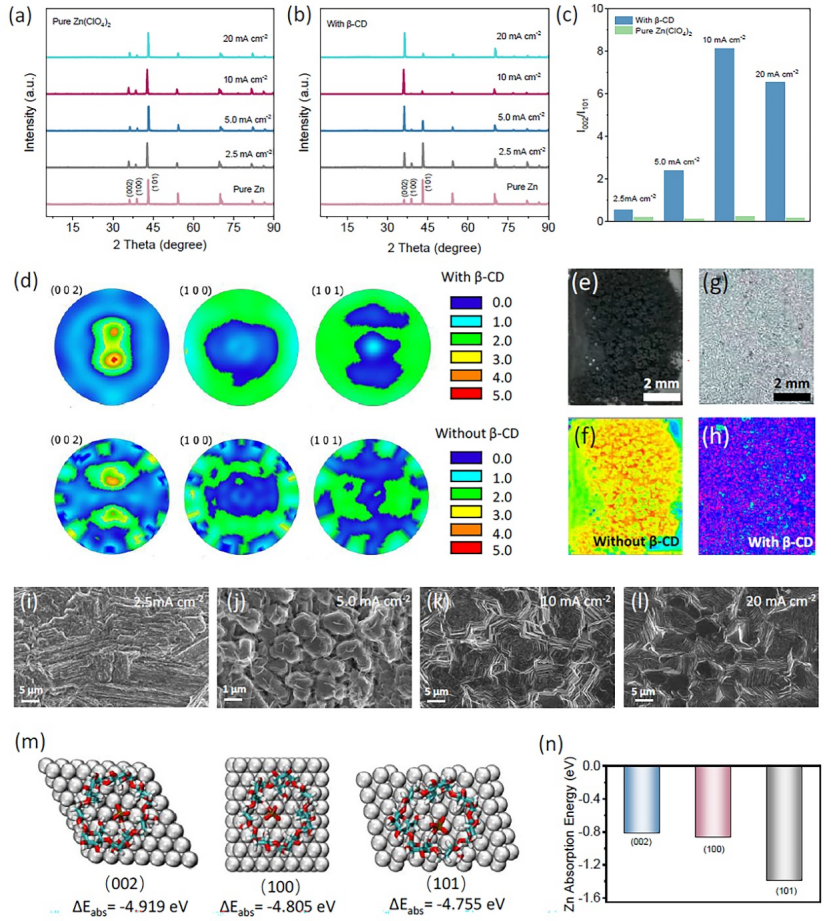
圖2. 鋅負極晶體重新取向的調節函數和機制。(a, b) 純Zn(ClO4)2和β-CD-Zn(ClO4)2電解質中Zn層的XRD圖譜;(c)不同電流密度下(002)和(101)晶面間強度比;(d) (002), (100), (101)沉積在Zn(ClO4)2-β-CD電解液中Zn負極(上)和商用Zn箔(下)的極圖;(e-h) 不同電解液Zn負極的光學照片;(i-l) 不同電流密度下Zn(ClO4)2-β-CD電解液中Zn負極SEM圖;(m) β-CD@ClO4-在Zn(002)、(100)和(101)表面及其相應Eabs上的復合吸附;(n) β-CD@ClO4-配合物吸附后,不同晶面的鋅原子吸收能。
隨后,研究了鍍鋅層的結構和形貌(固定面積容量為10 mAh cm-2,電流密度2.5~20 mA cm-2)。顯然,與原始Zn負極相比,Zn(002)的相對強度都得到增強,對于純Zn(ClO4)2體系,不同電流密度下R值幾乎沒有差異,意味著對特定Zn(002)的生長沒有修飾作用。相比之下,β-CD的加入會極大地影響R值隨沉積條件的變化,實現最好Zn(002)的電流密度應為10 mA cm-2。
隨后研究了不同電解質系統的Zn表面,顯然,由β-CD輔助的沉積Zn表現出強烈集中以Zn(002)分布,不同電流密度SEM圖像表明,添加β-CD可以有效地調節上述負極表面的Zn(002)組織織構,表明該策略具有普適性。
為了更好理解β-CD在促進優選晶體取向方面的潛在機制,對不同晶面上的幾種吸收行為進行初始計算,由布拉維定律可知,最終暴露出的晶面通常具有最低的生長速率。因此,為了促進Zn(002)面的更多暴露,應限制Zn2+沉積速度,計算發現β-CD@ClO4-配合物在(002)、(100)和(101)上吸收能(Eabs)分別為-4.919 eV、-4.805 eV和4.755 eV,所以其優先與Zn(002)結合。
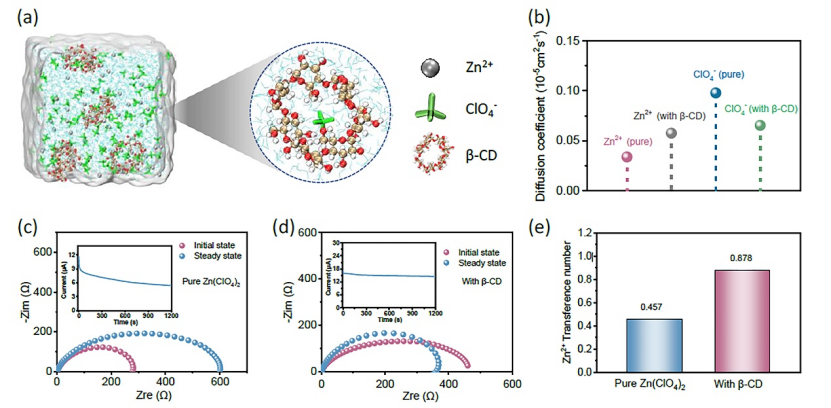
圖3. β-CD@ClO4-配合物Zn2+快速擴散理論模擬和電化學測試。(a) Zn(ClO4)2-β-CD電解液三維結構模擬;(b) MD模擬Zn2+和ClO4-的擴散系數;(c, d) 電化學阻抗譜;(e) Zn2+遷移數對比。
電解質環境和電極界面內的Zn2+遷移率是影響Zn負極穩定性和ZIBs整體性能的另一個重要因素。利用分子動力學(MD)模擬,探討陰離子和β-CD之間相互作用對Zn2+和陰離子傳輸動力學的影響,從模擬采集的Zn(ClO4)2-β-CD的放大圖可以看出,ClO4-明顯進入β-CD空腔,β-CD內腔會對ClO4-陰離子遷移施加空間限制,導致Zn2+-ClO4-離子對解耦,從而使β-CD改性電解質轉變為具有高流動性Zn2+受限狀態。
計算Zn2+遷移數(tZn2+)發現,Zn(ClO4)2-β-CD電解質為0.878,遠高于純Zn(ClO4)2,接近單離子傳輸電解質(tZn2+=1)。Zn(ClO4)2-β-CD電解質中Zn2+遷移活化能更低也表明Zn2+遷移能壘更低。
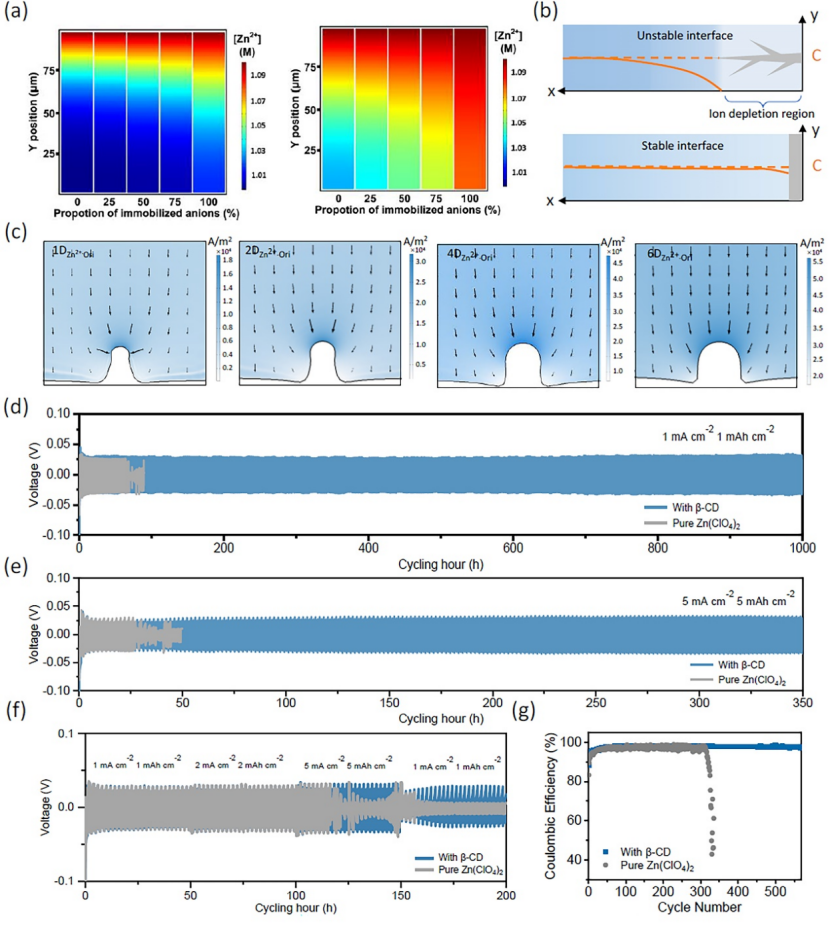
圖4. 鋅離子遷移行為分析及鋅沉積長期穩定性測試。(a) 1.0 s和(右)5.0 s后Zn2+從負極表面轉移到本體電解質(左);(b) 不同電解質中鋅沉積過程的枝晶生長情況;(c) 不同Zn2+擴散速率下Zn負極電流密度分布。(d, e) 不同電流密度和面積容量下,Zn||Zn對稱電池的長期循環性能比較;(f) 不同電流密度和面積容量下,Zn||Zn對稱電池倍率循環性能比較;(g)不同電解質Zn||Cu電池庫倫效率比較。
進一步通過有限元分析(FEA),來闡明遷移速率對離子濃度分布的影響。通常,均勻分布的Zn2+可以引導無枝晶Zn沉積。在沒有β-CD添加劑電解液中(固定陰離子比率低),容易引發枝晶生長,隨著陰離子固定化率的增加(Zn(ClO4)2-β-CD),Zn2+更容易從體電解質擴散到負極表面,從而形成均勻Zn2+分布環境,而不會出現枝晶。
考慮到Zn2+擴散系數,進一步模擬上述抑制枝晶生長行為,β-CD添加后在可能發生枝晶的位置出現更均勻電流密度分布,從而產生了填充和平整效果。以上實驗結果和有限元分析表明,β-CD的陰離子陷阱效應有利于更流暢的Zn2+轉移,Zn||Zn對稱電池長期循環進一步證明了β-CD對枝晶生長抑制的增強作用。
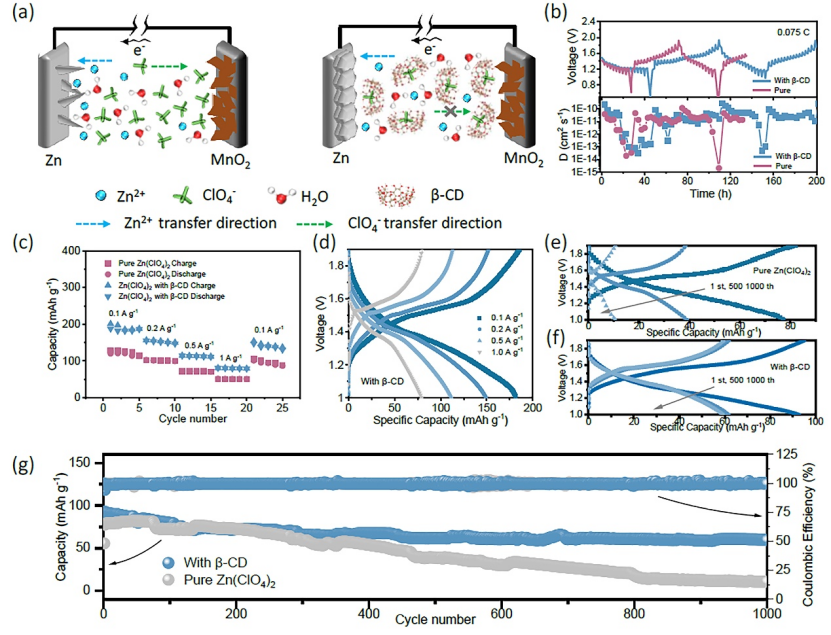
圖5. 不同電解質下Zn-MnO2全電池性能。(a) Zn-MnO2全電池充放電機理圖;(b) GITT測試及離子擴散系數計算;(c) 倍率性能;(d) Zn(ClO4)2-β-CD電解液下,Zn-MnO2全電池放電曲線;(e-g) 循環穩定性。
最后,為了證實β-CD內部陰離子阱的綜合性能,采用不同的Zn-MnO2全電池進行電化學性能測試。GITT結果表明,在具有β-CD的電解質體系中,Zn2+遷移速率比純Zn(ClO4)2電解質中更快(D值高約10倍)。倍率性能測試發現,引入β-CD添加劑后,Zn-MnO2電池都表現出明顯更高容量(增強了約51%至57%),其原因可以歸結為:(1)更高的Zn2+遷移數保證了電解質和電極/電解質界面之間穩定充足的Zn,使兩個電極側反應效率更高;(2)更好的Zn2+遷移行為有利于MnO2正極中Zn2+更快擴散。
同時發現,添加β-CD后循環穩定性更好,即使循環1000 次也能保持63.9%的初始容量,主要原因有:(1)陰離子阱(β-CD)保證Zn2+更好的可逆進出MnO2正極;(2)負極上優選Zn(002)的獨特調節作用,進一步優化整個全電池系統中Zn2+利用率,從而有助于提高穩定性。
總 結
作者首次提出了一種“陰離子陷阱”策略,即通過使用β-CD作為ClO4-的主體劑來提高鋅離子電池性能。通過理論計算、光譜表征和ITC測量明確證明了β-CD疏水內腔與ClO4-之間的主客體相互作用,及β-CD@ClO4-配合物的形成。由于固定化ClO4-的均勻分散,Zn2+遷移數從0.457提高到0.878,抑制了無規枝晶生長。同時,β-CD@ClO4-配合物通過控制不同晶面生長速率,抑制鋅枝晶出現,使Zn(002)晶面優先暴露。以上協同作用導致Zn||Zn對稱電池和Zn-MnO2全電池性能顯著提升。這項工作為電解質設計和實現高性能水系電池提供了全新見解。
審核編輯:劉清