背景介紹
固體聚合物電解質(SPEs)在固態鋰電池中有著廣闊的應用前景,但目前廣泛應用的PEO基聚合物電解質室溫離子電導率和機械性能較差,電極/電解質界面反應不受控制,限制了其整體電化學性能。雖然PEO基電解質的室溫鋰離子電導率可以通過添加惰性或活性填料來改善,但其機械性能和電化學穩定性仍然不足,限制了全電池的能量密度和循環壽命。
與PEO相比,PVDF基電解質具有較高的機械強度、良好的熱穩定性和較寬的電化學窗口。然而,如何在不過度增加厚度的情況下有效提高聚合物電解質的力學性能,構建穩定的電極/電解質界面層仍是實現高能量密度PVDF基固態鋰金屬電池需要解決的關鍵問題。
正文部分
1、成果簡介
近日,青島大學郭向欣教授和北京航空航天大學宮勇吉教授團隊,利用二維氟化石墨烯增強PVDF-HFP-LiTFSI(FPH-Li)聚合物電解質以實現高性能固態鋰金屬電池。均勻分散的氟化石墨烯能夠誘導晶粒細化,在不過度增加聚合物電解質厚度的情況下有效提高了其機械性能。
聚合物晶粒尺寸的顯著減小增強了界面鋰離子的傳輸并使鋰離子分布均勻,從而提高了鋰離子電導率并促進了均勻的鋰電鍍/剝離。此外,大量表征表明,氟化石墨烯參與構建了穩定的人工界面層,有效防止了鋰金屬負極與溶劑化分子之間的副反應。
因此,使用薄的FPH-Li聚合物電解質(厚度約為45 μm)可以在Li/Li對稱電池中以較小的過電勢實現穩定的Li 電鍍/剝離。用其組裝的Li/LiNi0.6Co0.2Mn0.2O2全電池能夠在1.0 C下穩定循環,平均庫侖效率高達99.5%。

2、研究亮點
本工作報道了一種二維(2D)氟化石墨烯增強PVDF-HFP-LiTFSI固體聚合物電解質(FPH-Li)的策略,該電解質具有更高的機械強度、較高的室溫鋰離子電導率和優異的電極/電解質界面穩定性。這種FPH-Li電解質涉及的幾個優點有利于提高綜合性能(圖1b)。
首先,二維氟化石墨烯的引入為PVDF-HFP的結晶提供了足夠的成核位點,并限制了PVDF-HFP晶粒的生長過程,從而減小了聚合物顆粒的尺寸。晶粒尺寸的減小引起了與金屬基復合材料類似的細晶強化效應,有效提高了FPH-Li電解質的機械強度。其次,FPH-Li薄膜的界面面積隨著聚合物粒徑的減小而增加。
均勻分散的氟化石墨烯在一定程度上抑制了聚合物晶粒的快速生長,從而改善了PVDF-HFP的鏈段運動,提高了室溫鋰離子電導率,并使鋰離子流均勻化。另外,氟化石墨烯表面的含氟基團與鋰金屬反應生成致密、穩定的人工界面層,有效防止電極與FPH-Li電解質之間的副反應。
?
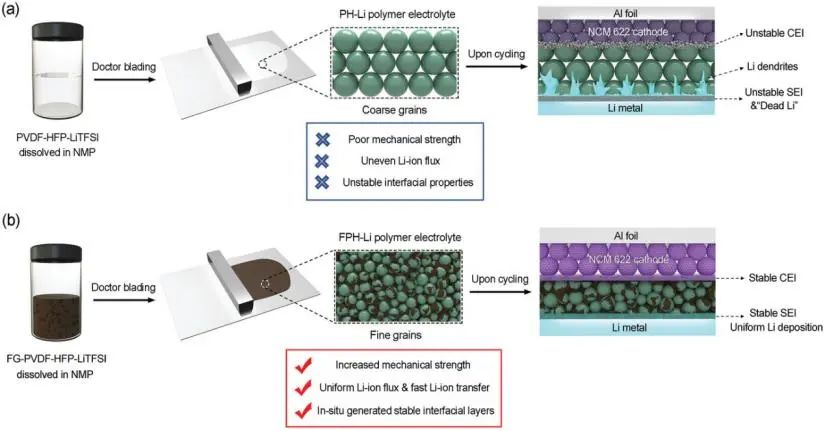
【圖1】a)普通PVDF-HFP-LiTFSI(PH-Li)聚合物電解質的合成過程及其固有缺陷對全電池循環的影響。b)氟化石墨烯增強PVDF-HFP-LiTFSI(FPH-Li)聚合物電解質的合成工藝及其對提高全電池循環穩定性的作用機理。
3、圖文導讀
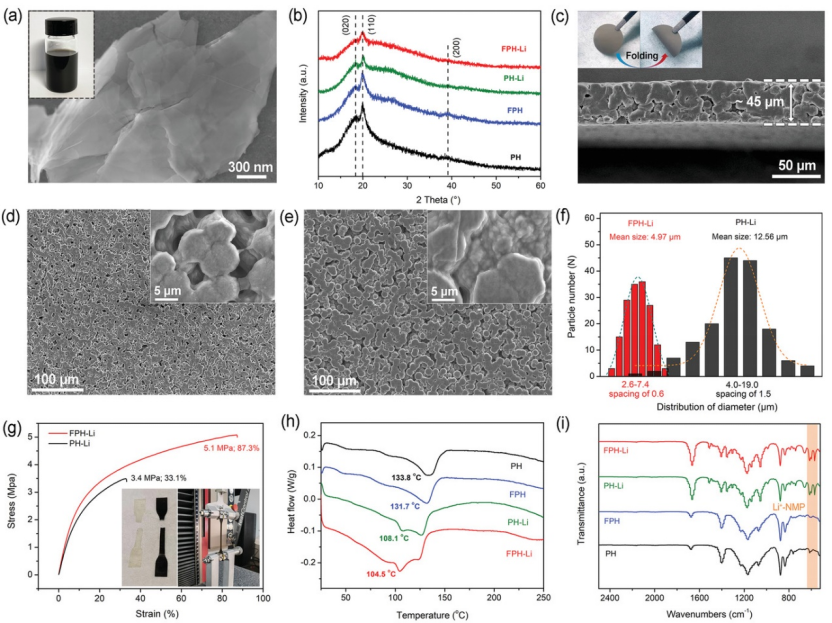
【圖2】a)氟化石墨烯的SEM圖像。(b)FPH-Li、PH-Li、FPH和PH聚合物膜的XRD圖譜 c)FPH-Li的橫截面SEM圖像。d)FPH-Li的SEM圖像。e)PH-Li的SEM圖像。f)FPH-Li和PH-Li電解質粒徑分布統計圖。g)FPH-Li和PH-Li電解質在拉伸試驗期間的應力-應變曲線。FPH-Li、PH-Li、FPH和PH聚合物膜的h)DSC曲線和i)FT-IR曲線。
氟化石墨烯(FG)是通過以氟化石墨為前驅體、NMP為插層分子的插層剝離策略獲得的。圖2a顯示,具有層狀結構的FG均勻分散在NMP中。
FPH-Li和PH-Li聚合物電解質膜是通過簡便的流延法制備的。圖2b顯示,PVDF-HFP(PH)和氟化石墨烯修飾的PVDF-HFP(FHP)聚合物膜在不添加鋰鹽的情況下,分別在18.5°、20.0°和39°處出現了對應α相PVDF-HFP(020)、(110)和(200)面的三個特征峰,沒有檢測到LiTFSI的衍射峰,表明聚合物電解質膜中的鋰鹽完全溶解。FPH-Li和PH-Li中PVDF-HFP對應的衍射峰強度顯著降低,這可歸因于LiTFSI降低了聚合物基體的結晶度。
此外,與PH-Li相比,FPH-Li中PVDF-HFP衍射峰的寬度增加,強度減弱,表明其晶粒尺寸較小,結晶度較低,有助于提高鋰離子電導率。掃描電子顯微鏡(SEM)顯示,FPH-Li電解質的厚度約為45 μm(圖2c),使其具有出色的柔性。
圖2d-e顯示,相比于PH-Li電解質松散的結構,FPH -Li電解質的晶粒較小且密集堆積,形成連續的織構。高倍SEM圖像顯示,在FPH-Li電解質表面出現了薄的FG,而在PH-Li電解質中沒有。FG不僅可以橋接聚合物顆粒以限制其快速生長,還可以構建大量的FG/PVDF-HFP接觸界面,以提高FPH-Li電解質的鋰離子傳輸速率。
此外,粒度統計結果(圖2f)表明,FPH-Li電解質的平均粒度約為4.97 μm,遠小于PH-Li電解質(≈12.56 μm)。粒徑的減小引起了細晶強化效應,極大地提高了FPH-Li電解質的機械性能。因此,FPH-Li電解質的斷裂強度和總伸長率分別為5.1 MPa和87.3%,遠優于3.4 MPa和33.1%的PH-Li電解質(圖2g)。圖2h顯示,PH、FPH、PH-Li和FPH-Li膜的玻璃化轉變溫度(Tg)分別為133.8、131.7、108.1和104.5°C,表明由于TFSI-的增塑作用,LiTFSI可以大大降低PVDF-HFP結晶度。
此外,FPH-Li的Tg略低于PH-Li,說明氟化石墨烯的引入不僅可以誘導成核,減小晶粒尺寸,還可以略微降低聚合物的結晶度。此外,FPH-Li電解質的傅里葉變換紅外(FT-IR)光譜(圖2i)顯示,在1295、1262、1110、983、926、655和470 cm-1處沒有游離NMP的特征峰,表明所有NMP分子都與Li+配位,沒有游離的NMP。
位于570、616、1055和1352 cm-1處的特征峰對應溶劑化的[Li(NMP)x]+分子。這表明FPH-Li電解質中的NMP以結合態而非游離態存在。溶劑化[Li(NMP)x]+在界面處的快速傳輸導致FPH-Li聚合物電解質具有優異的鋰離子電導率。
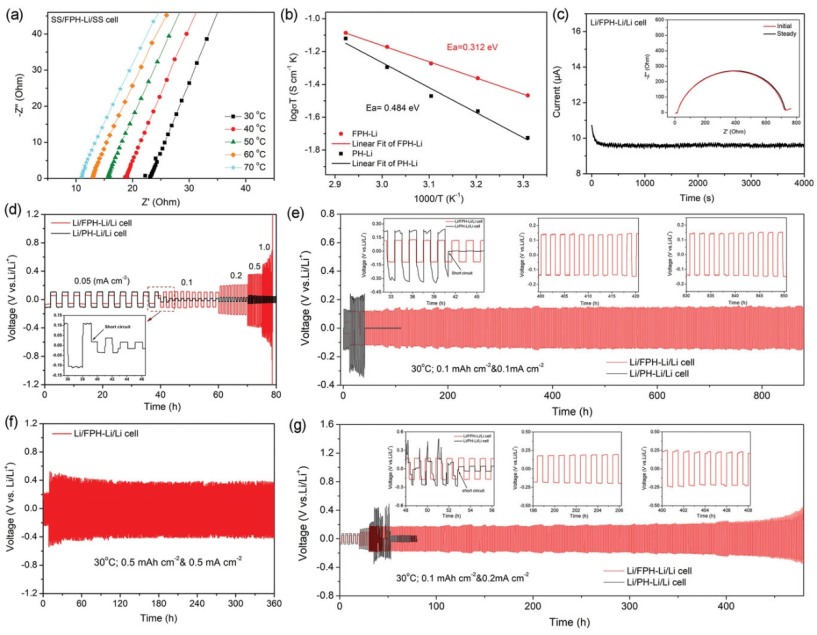
【圖3】a)SS/FPH-Li/SS電池在不同溫度下的奈奎斯特圖。b)FPH-Li和PH-Li電解質的離子電導率Arrhenius圖。c)Li/FPH-Li/Li電池在10 mV極化下的計時電流曲線。d)Li/FPH-Li/Li和Li/PH-Li/Li電池在不同電流密度下的恒流充放電曲線。e)Li/FPH-Li/Li和Li/PH-Li/Li電池在0.1 mA cm-2下1 h的恒流循環曲線。f)Li/FPH-Li/Li電池在0.5 mA cm-2下1 h的恒流循環曲線。g)Li/FPH-Li/Li和Li/PH-Li/Li電池在0.2 mA cm-2下1 h的恒流循環曲線。測試溫度為30 °C。
使用不銹鋼(SS)作為阻塞電極組裝對稱電池,以測量固體聚合物電解質的鋰離子電導率。根據電化學阻抗譜(EIS),可以計算出FPH-Li電解質在30 °C的鋰離子電導率為1.32 × 10-4?S cm-1(圖3a),遠高于PH -Li電解質(6.21 × 10-5?S cm-1)。不同溫度下鋰離子電導率的Arrhenius圖(圖3b)表明,FPH-Li和PH-Li電解質的鋰離子遷移活化能(Ea)分別為0.312和0.484 eV,表明FPH-Li聚合物電解質中的鋰離子傳輸勢壘較低。
圖3c顯示,FPH-Li的鋰離子遷移數(tLi+)為0.472,高于PH-Li(tLi+=0.315)。DC極化的結果表明,FPH-Li電解質的電子電導率為 1.98 × 10-9?S cm-1,表明氟化石墨烯的引入不會增加電子電導率。
圖3d顯示,Li/FPH-Li/Li的極化電壓隨著電流密度的增加而增加,即使在1.0 mA cm-2下,電池也能保持穩定的循環而不發生短路。相比之下,當電流密度增加到0.1 mA cm-2時,Li/PH-Li/Li對稱電池發生短路,這可歸因于不穩定的界面和嚴重的鋰枝晶生長。
Li/FPH-Li/Li對稱電池在0.1 mA cm-2@30 °C下能夠穩定循環超過900小時,而Li/PH-Li/Li對稱電池電池僅在40小時后短路。當電流密度增加到0.2 mA cm–2時,Li/FPH-Li/Li對稱電池可以穩定運行超過500小時(圖3g)。而Li/PH-Li/Li對稱電池電壓曲線波動劇烈,電壓極化大,電池迅速短路。即使在0.5 mA cm-2@0.5 mAh cm-2下循環,Li/FPH-Li/Li對稱電池也可以運行360小時(圖3f)。
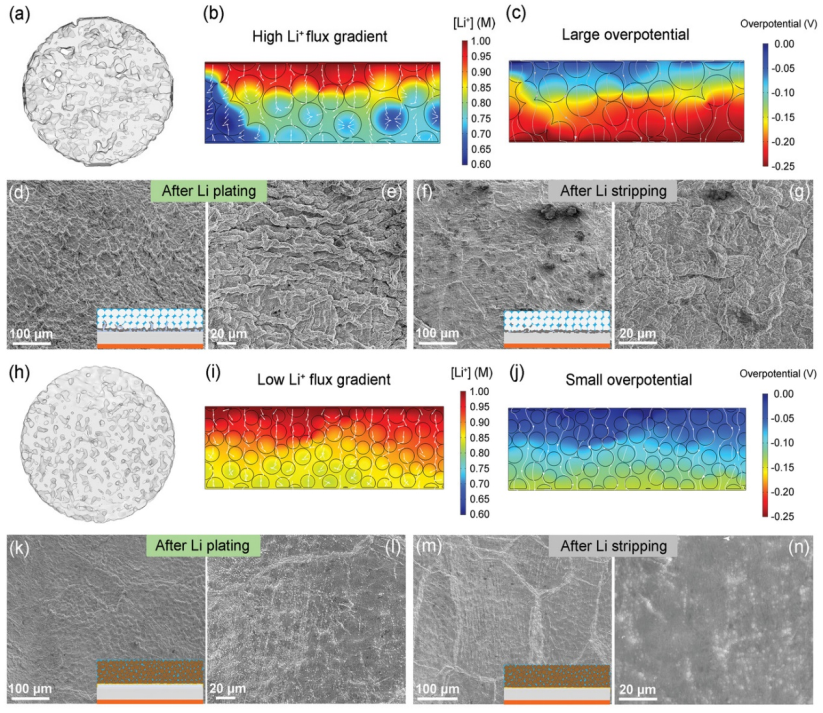
【圖4】a)PH-Li電解質的水平切片同步X射線計算機斷層掃描(SX-CT)圖。COMSOL模擬了PH-Li電解質中b)平衡鋰離子濃度剖面和c)過電位分布。Li/PH-Li/Li電池在0.1 mA cm-2下d-e)電鍍0.1 mAh cm-2和f-g)剝離0.1 mAh cm-2后Li金屬負極的SEM圖像。h)FPH-Li電解質水平切片SX-CT圖。COMSOL模擬FPH-Li電解質中的i)平衡鋰離子濃度剖面和j)過電位分布。Li/FPH-Li/Li電池在0.1 mA cm-2下k-l)電鍍0.1 mAh cm-2和m-n)剝離0.1 mAh cm-2后Li金屬負極的SEM圖像。
為了揭示氟化石墨烯提高聚合物電解質對鋰金屬穩定性的機制,對鋰金屬的電鍍/剝離行為進行了表征。首先使用同步加速器X射線計算機斷層掃描(SX-CT)表征不同電解質膜的孔隙率和界面特性。圖4a顯示,由于PVDF-HFP具有大的粒徑,在PH-Li電解質中可以觀察到許多大的空隙,這會惡化界面接觸并阻礙界面鋰離子傳輸。
通過使用有限元方法求解離子擴散和靜電勢場,進一步模擬聚合物電解質中鋰離子濃度分布的動力學平衡。圖4b顯示,由于聚合物顆粒的大尺寸,PH-Li電解質膜的表面和內部形成了大的空隙。這些空隙阻礙了鋰離子的傳輸,導致PH-Li電解質中的鋰離子分布不均勻。PH-Li電解質中的高鋰離子濃度梯度將導致鋰電鍍/剝離產生高的過電勢(≈0.25 V,圖4c),這會誘導快速的鋰枝晶生長。
圖4d顯示,在以0.1 mA cm-2電鍍0.1 mAh cm-2的Li后,可以在電極表面觀察到巨大的鋰沉積物。放大的SEM圖像(圖4e)表明鋰沉積物呈枝晶狀。在鋰剝離后,大量的“死鋰”留在電極表面(圖4f-g),進一步惡化了PH-Li電解質/鋰界面的穩定性。
相比之下,FG引入帶來的晶粒細化效應大大降低了FPH-Li聚合物電解質的粒徑,不僅增強了界面接觸,還減少了大空隙的產生。水平切片SX-CT圖像(圖4h)顯示,FPH-Li電解質的孔隙率大大降低,界面接觸緊密。此外,鋰離子均勻分布在FPH-Li電解質中,有效降低了鋰離子濃度梯度(圖4i)。
同時,鋰電鍍/剝離的過電位降低(≈0.1 V,圖4j),有助于抑制鋰枝晶的形成。Li/FPH-Li/Li電池的SEM圖像(圖4k-l)顯示,在以0.1 mA cm-2電鍍0.1 mAh cm-2的Li后,電極表面均勻平整,沒有鋰枝晶。在鋰剝離后,電極仍然呈現光滑的表面,沒有觀察到“死鋰”(圖4m-n)。
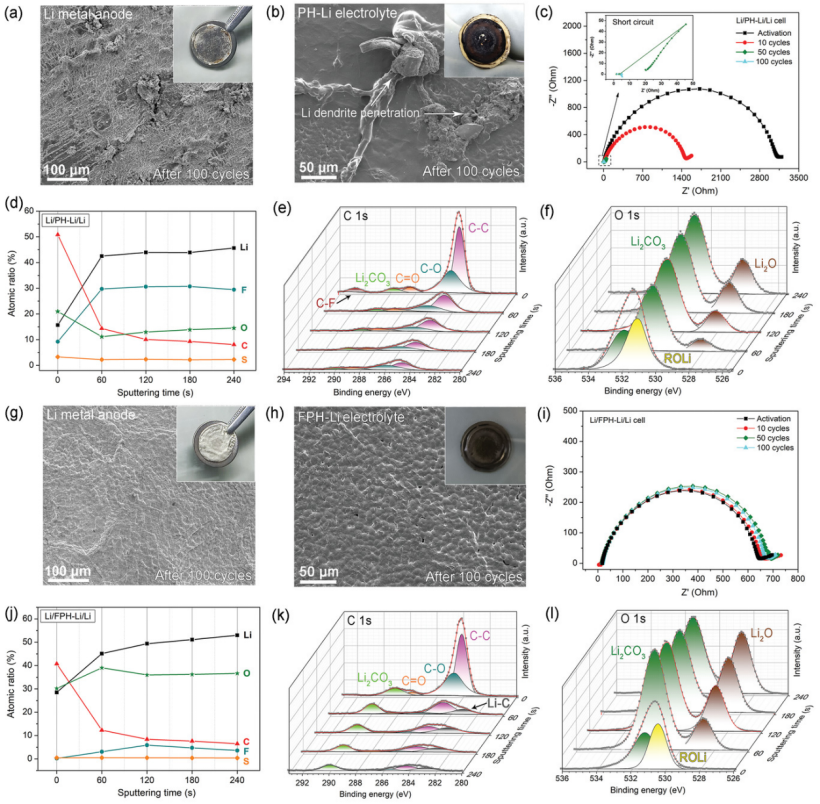
【圖5】Li/PH-Li/Li電池第100次Li剝離后,a)Li負極的SEM圖像和照片,b)PH-Li電解質的SEM圖像和照片。c)Li/PH-Li/Li電池在不同循環下的EIS光譜。d)不同濺射時間后Li/PH-Li/Li電池中循環鋰金屬負極的原子組成比例和相應的e)C1s和f)O1s光譜。Li/FPH-Li/Li電池第100次Li剝離后,g) Li負極的SEM圖像和照片,h)FPH-Li電解質的SEM圖像。i)Li/FPH-Li/Li電池在不同循環下的EIS光譜。j)不同濺射時間后Li/FPH-Li/Li電池中循環鋰金屬負極的原子組成比例和相應的k)C1s和l)O1s光譜。
圖5a顯示,經過100次鋰電鍍/剝離循環后,Li/PH-Li/Li電池中的電極表面變得粗糙和暗淡,并帶有大量的“死鋰”。循環后的PH-Li電解質膜變成深棕色(圖5b),在PH-Li電解質的表面上積累了大量的鋰枝晶和副反應產物。EIS光譜顯示,50次循環后界面電阻急劇下降,表明Li/PH-Li/Li電池發生了微短路(圖5c)。
通過X射線光電子能譜(XPS)進一步確定了100次循環后鋰金屬負極上SEI層的組成。圖5d顯示,F的含量在濺射時從9.21%迅速增加到29.76 at%,并在整個濺射過程中保持在30at% 以上。如此高含量的LiF表明,PH-Li電解質中的PVDF-HFP發生了不可控的脫氟化氫反應,導致形成具有多孔結構的厚SEI層。
濺射前C的含量高達50.84%,在整個氬離子刻蝕過程中C和O的含量都保持在較高水平。詳細的C1s和O1s光譜(圖5e-f)表明,與SEI層中的有機組分相對應的C-O和C=O鍵的比例增加,表明鋰金屬負極和溶劑化分子之間的副反應加劇。
相比之下,即使經過100次循環,Li/FPH-Li/Li電池的電極表面仍然相對平坦,沒有鋰枝晶或死鋰的積累(圖5g)。FPH-Li電解質表面原位生成穩定的界面層,100次循環后仍保持均勻光滑。在FPH-Li電解質中沒有觀察到鋰枝晶穿透(圖5h)。EIS光譜顯示,Li/FPH-Li/Li電池的界面電阻在不同的循環中保持不變(圖5i),表明界面穩定。圖5j顯示了在不同XPS刻蝕深度下,Li金屬電極(100次循環后從Li/FPH-Li/Li電池中提取)上特定元素的原子比。F含量在濺射120秒后達到最高值(5.88 at%)。
如此低的F含量表明LiF不是源自FPH-Li電解質膜中PVDF-HFP的脫氟化氫反應,而是由氟化石墨烯和鋰金屬之間的原位反應生成。C含量從40.72下降到8.37 at%。C 1s光譜(圖5k)表明,C-C和Li2CO3是主要成分,而初始階段出現的C-O和C=O鍵對應于SEI中的有機組分。
此外,在濺射60 s后,Li/PH-Li/Li電池中Li-C鍵的出現可歸因于鋰離子與石墨烯的相互作用。Li-C鍵的出現進一步表明氟化石墨烯參與了SEI層的構建。隨著濺射時間的增加,Li和O的含量(圖5l)不斷增加并成為主要元素,表明Li金屬均勻地沉積在SEI層下。
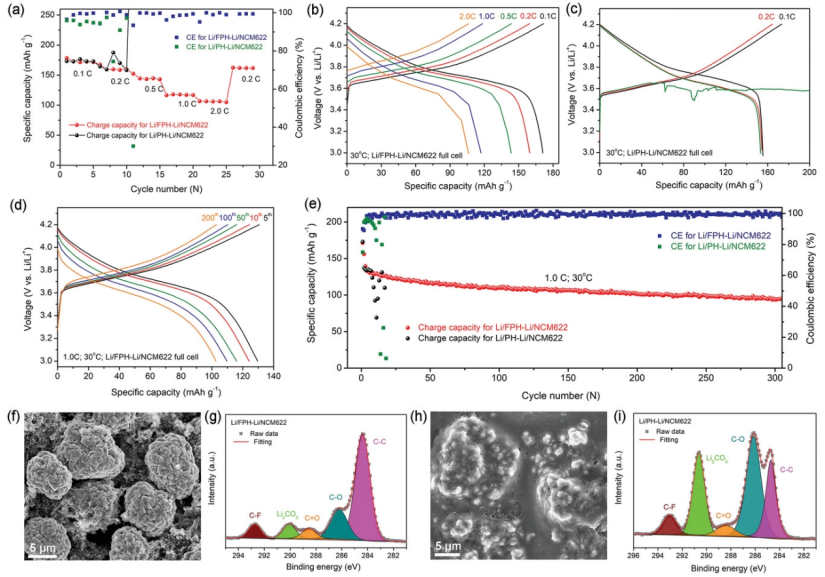
【圖6】a)Li/FPH-Li/NCM622和Li/PH-Li/NCM622電池的倍率性能。b)Li/FPH-Li/NCM622和c)Li/PH-Li/NCM622電池在不同倍率下的充放電曲線。d)Li/FPH-Li/NCM622在1.0 C下的充放電曲線。e)Li/FPH-Li/NCM622和Li/PH-Li/NCM622電池在1.0 C下的循環性能。1.0 C下循環20次后,在Li/FPH-Li/NCM622中NCM622正極的f)SEM圖像和g)XPS C1s光譜。1.0 C下循環20次后,Li/PH-Li/NCM622中NCM622正極的h)SEM圖像和i)XPS C1s光譜 。
進一步組裝了使用FPH-Li和PH-Li電解質的Li/NCM622全電池,并在30°C下進行了測量,以驗證FPH-Li電解質的實用性。圖6a顯示,Li/FPH-Li/NCM622電池在0.1、0.2、0.5、1.0和2.0 C下分別提供173.1、159.7、144.8、118.0和106.3 mAh g-1的容量,當電流密度恢復到0.2 C時,容量仍有159.3 mAh g-1,證明Li/FPH-Li/NCM622電池具有優異的倍率性能。
Li/FPH-Li/NCM622電池在不同倍率下的充放電曲線是穩定的,表明沒有發生副反應(圖6b)。而Li/PH-Li/NCM622電池在0.2C時容量波動劇烈,0.5C時全電池失效。在充電/放電曲線(圖6c)中,對應于副反應的電壓平臺出現在3.7 V左右。圖6d-e顯示,Li/FPH-Li/NCM622電池可在1.0 C下穩定循環300多次,容量保持率約為81.5%,平均庫侖效率為99.5%,而Li/PH-Li/NCM622電池容量快速衰減,電池僅在15個循環后失效(圖6e)。
SEM(圖6f)表明,使用FPH-Li電解質的NCM622電極表面仍然整潔光滑,沒有厚的涂層。然而,對于使用PH-Li電解質的NCM622電極,電極顆粒被厚涂層包裹(圖6h)。循環后NCM622電極的C1s(圖6g)XPS光譜顯示,對于Li/FPH-Li/NCM622全電池中的NCM622電極,對應于正極電解質界面(CEI)的C-O、C=O和poly(CO3)峰強度相對較低,表明電解質的分解有限,FPH-Li電解質/正極界面穩定。
相比之下,對于使用PH-Li電解質的NCM622電極,可以觀察到與CEI對應的C-O、C=O 和poly(CO3)峰強度顯著增加,表明PH分解嚴重,PH-Li電解質/正極界面不穩定(圖6i)。
4、總結和展望
本工作設計了一種新的二維氟化石墨烯增強PVDF-HFP固體聚合物電解質,用于室溫鋰金屬電池。添加氟化石墨烯引起的晶粒細化效應不僅可以改善機械性能,還可以增強界面鋰離子傳輸,從而提高聚合物電解質的鋰離子電導率。此外,氟化石墨烯參與構建穩定的界面層,提高了鋰金屬負極和NCM622正極側聚合物電解質的電化學穩定性,實現了固態Li/FPH-Li/NCM622全電池的長循環。
本工作驗證了二維材料在提高聚合物電解質綜合性能方面的可行性,為推動基于固體聚合物電解質的SSLBs發展提供了新思路。
審核編輯:劉清