01 ? 導讀
水系鋅離子電池(ZIBs)因其高容量和優異的安全性而有望成為下一代儲能技術。然而,鋅金屬負極在循環過程中會形成枝晶并發生析氫副反應,從而導致電池庫侖效率(CE)較低,電極過早失效。對Zn2+溶劑化鞘層進行調控是抑制析氫反應(HER)副反應最直接的方法。但調節溶劑化鞘層會導致Zn2+成核/溶解動力學較差,嚴重影響Zn電鍍/剝離可逆性。因此,如何解決鋅成核/溶解動力學與HER抑制能力之間的內在權衡對于開發安全高效的鋅離子電池至關重要。
02 ? 成果背景
近日,Angew. Chem. Int. Ed.上發表了一篇題為“Regulating Surface Reaction Kinetics through Ligand Field Effects for Fast and Reversible Aqueous Zinc Batteries”的文章,該文章發現具有中等配體場相互作用的硼酸(BA)可以部分取代Zn2+溶劑化鞘中的H2O分子,形成穩定的缺水溶劑化鞘。它能夠實現快速的Zn成核/溶解動力學并顯著抑制HER。通過系統地比較BA和其他電解質添加劑的配體場強和溶劑化能,發現溶劑化能與Zn成核/溶解動力學和HER抑制能力有很強的相關性。該工作為鋅電池溶劑化鞘的設計提供了參考。
03 ? 關鍵創新
(1)BA可以部分取代Zn2+第一配位層中的水分子,形成新的缺水溶劑化鞘。
(2)原位光學顯微鏡和有限元分析(FEA)表明,BA還可以調節Zn電極表面周圍的電場,以實現均勻的Zn沉積。
(3)具有BA添加劑的Zn//Zn對稱電池可以在0.5 mA cm-2下穩定循環超過3000小時。即使在5 mA cm-2下,它也能提供84 mV的超低電壓極化和5.25 Ah cm-2的累積電鍍容量(CPC),表明Zn2+成核/溶解動力學顯著改善。
04 ? 核心內容解讀
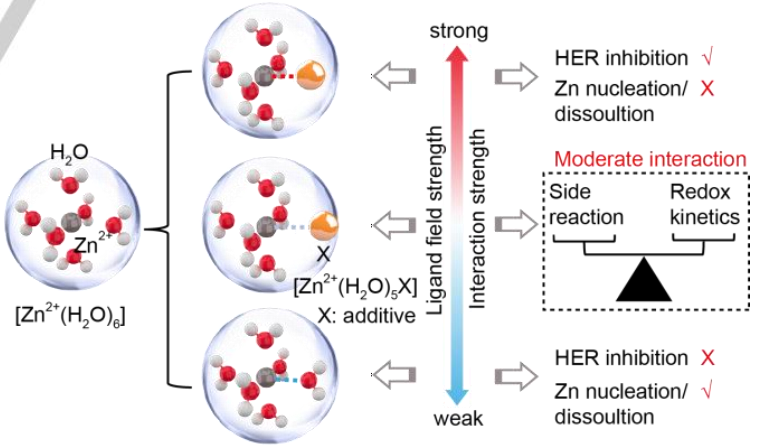
圖1.?Zn2+和添加劑之間的配體場強、相應的溶劑化鞘層以及它們對HER和Zn成核/溶解反應動力學的影響。@Wiley
基于經典配體場理論和Sabatier原理,尋找合適的配體,進而調節配體場強度和相應的氧化還原動力學在理論上是可行的。配體場強與副反應動力學的關系如圖1所示。其中配位作用過弱的配體不能穩定缺水的溶劑化鞘,導致有害的HER副反應,而過于穩定的溶劑化鞘則導致解溶劑化過程受阻,成核/溶解動力學遲緩。
因此,需要尋找具有適度配體場效應的新型電解質添加劑來平衡副反應和氧化還原動力學之間的矛盾。硼酸(BA)具有比大多數有機小分子更寬的氧化電位和略弱的配位能力,由于形成反饋π鍵,預計其與鋅離子具有適度的配體場相互作用。
?
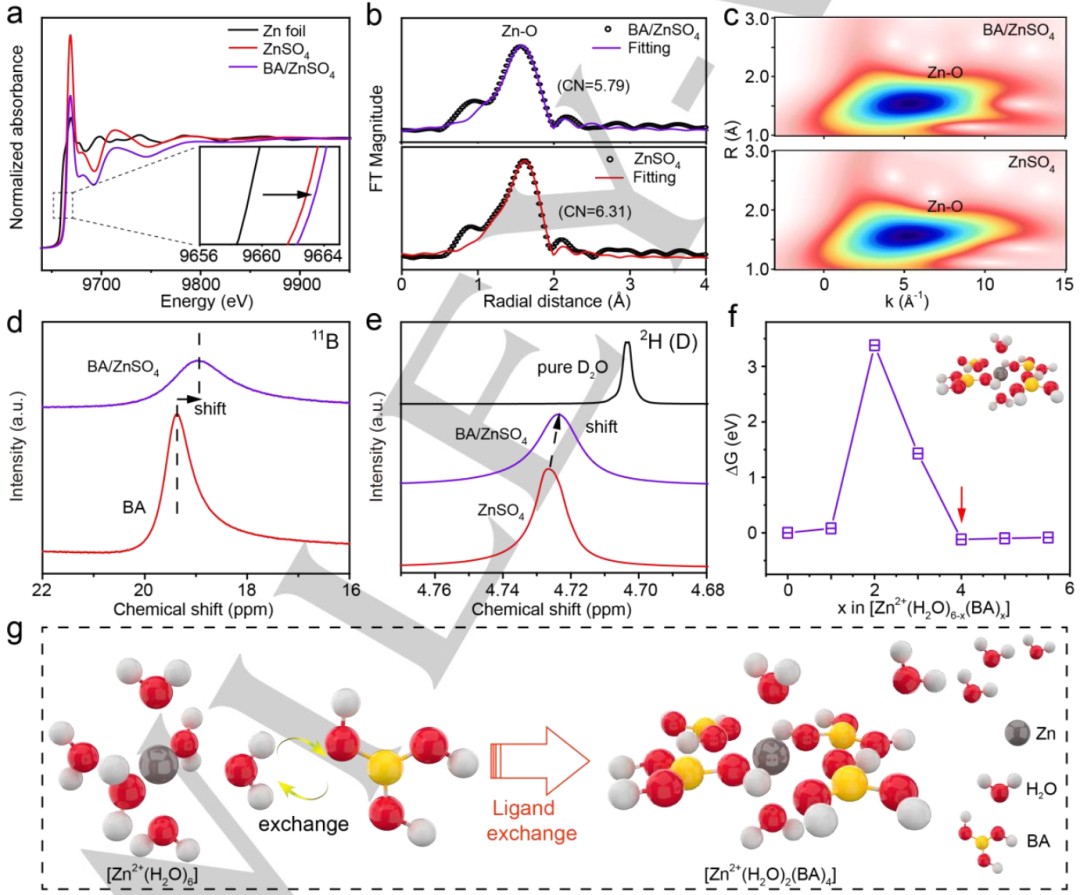
圖2.?a)歸一化的Zn K邊XANES譜。插圖:放大的Zn K邊XANES譜。b)R空間中的EXAFS譜。c)EXAFS譜的小波變換圖像。d)BA/ZnSO4電解液和BA溶液的11B NMR譜。e)BA/ZnSO4電解液、ZnSO4電解液和純D2O中的2H NMR譜。f)所有可能的Zn2+溶劑化鞘的ΔG。插圖是穩定的[Zn2+(H2O)2(BA)4]溶劑化鞘。g)引入BA后Zn2+溶劑化鞘的配體交換過程示意圖。@Wiley
為了揭示BA添加劑對Zn2+溶劑化鞘層的影響,進行了XAFS、拉曼光譜和 NMR測試。圖2a顯示了BA/ZnSO4電解質、Zn箔和ZnSO4電解質的Zn K邊X射線吸收近邊結構(XANES)譜。由于其固有的金屬特性,鋅箔具有最小的吸收邊能量。ZnSO4放大的Zn K邊XANES曲線顯示,在引入BA后,近邊吸收能量轉移到高能區,表明BA/ZnSO4電解質中Zn2+周圍的平均電子密度低于ZnSO4電解液。
此外,利用Zn K邊XANES譜在R空間的傅里葉變換(FT-EXAFS)探究BA/ZnSO4電解質和ZnSO4電解質的局部溶劑化鞘(圖2b)。~1.60 ?處的單個第一配位殼來源于Zn-O矢量。BA/ZnSO4電解液和ZnSO4電解液中Zn-O徑向距離分別為2.05和2.08 ?,Zn-O的配位數(CN)分別為5.79和6.31。此外,小波變換(WT)EXAFS用于分析Zn-O配位(圖2c)。CN和WT最大值相似,說明引入BA后仍為Zn-O配位。
此外,進行核磁共振譜以評估Zn2+和BA分子之間的相互作用。圖2d顯示,BA水溶液的11B峰位于19.372 ppm。添加2.0 M ZnSO4后,11B峰移至18.933 ppm,表明BA分子中B原子周圍電子密度增加,屏蔽效應增強。這種現象可以歸因于BA分子在Zn2+溶劑化鞘層中的取代。
此外,在核磁共振譜中,純D2O的2H峰通常位于4.703 ppm(圖2e)。引入2.0 M ZnSO4后,2H峰升高,達到4.727 ppm,說明2H的電子密度增加是由于Zn2+和D2O的配位作用導致。如果進一步向體系中加入BA分子,水的2H峰開始向前移動到4.723 ppm,表明一些被束縛的水分子再次被釋放。
為了進一步探究溶劑化鞘結構,進行了理論計算。考慮了各種BA和H2O比例下所有可能的溶劑化鞘結構,并計算了相應的吉布斯自由能(ΔG),如2f所示。根據計算結果,BA由于ΔG最低,容易取代4個水分子形成新的缺水溶劑化鞘,表明配體交換在熱力學上是有利的。這些結果說明BA添加劑通過配體交換改變了溶劑化鞘,如圖2g所示。
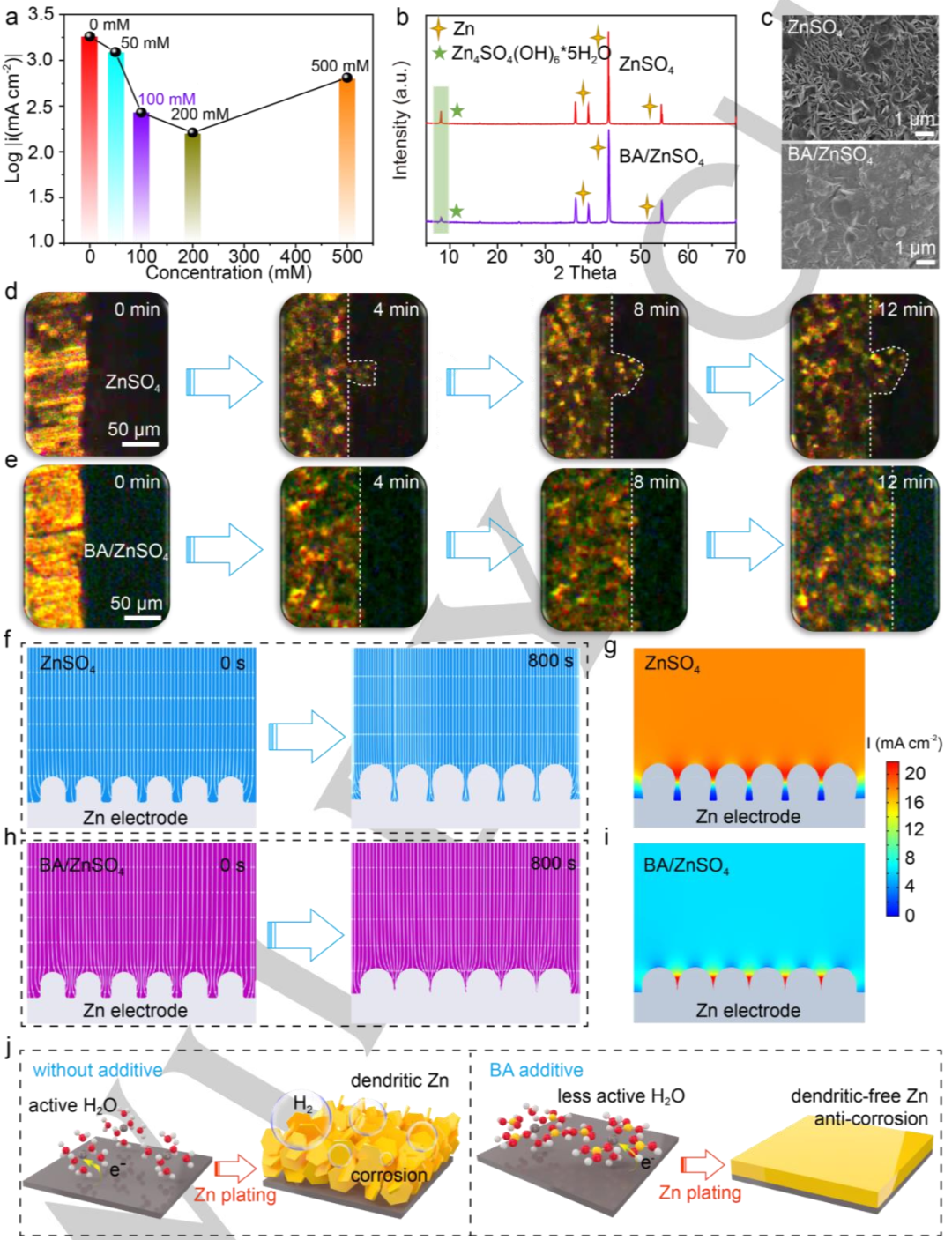
圖3.?a)不同溶液下的腐蝕電流密度。具有ZnSO4和BA/ZnSO4電解質的Zn//Cu電池循環50次后,Zn電極的b)XRD譜和c)SEM圖。d)ZnSO4,e)BA/ZnSO4電解質中,鋅沉積行為的原位光學顯微鏡研究。f,g)ZnSO4電解質,h,i)BA/ZnSO4電解質中,電極/電解質界面的FEA和相應的電流密度分布。j)BA添加劑在ZnSO4電解質中對Zn負極界面反應的改性機理示意圖。@Wiley
圖3a顯示,含有BA添加劑的電解質表現出較低的腐蝕電流密度,表明鋅負極的耐腐蝕性能得到增強。此外,利用X射線衍射(XRD)對在Zn//Cu電池中經過50次電鍍/剝離循環后的Zn電極進行表征(圖3b)。在8.1°附近的峰,與副產物Zn4SO4(OH)6·5H2O相關,在添加BA后,該峰強度顯著降低,再次證實了BA對腐蝕的抑制作用。
圖3c的掃描電子顯微鏡(SEM)圖像顯示,由于鋅與水系電解質之間的連續反應,沒有BA添加劑的鋅循環后,表面變得粗糙,存在片狀枝晶。相比之下,在BA/ZnSO4電解液中循環的Zn表現出致密和光滑的形貌。因此,BA添加有望調節鋅沉積行為。
為了進一步研究溶劑化鞘對鋅沉積行為的影響,采用原位光學顯微鏡直接觀察電解質-電極界面處的鋅沉積形貌。對于沒有BA添加劑的Zn電極,在40 mA cm-2下沉積4分鐘后會出現不均勻的成核位點(圖3d)。這些突出的鋅位點不受控制地長成枝晶,最終導致循環可逆性差和電池失效。相反,使用BA添加劑,沉積過程看起來均勻且穩定(圖3e)。電鍍12分鐘后,沒有觀察到類似枝晶的形貌,表明添加BA有利于使成核位點均勻化,最終抑制了Zn枝晶的生長。
有限元分析(FEA)用于進一步了解了BA添加劑對Zn沉積行為的影響。圖3f顯示,在沒有BA添加劑的情況下,Zn的連續沉積往往會發生在不平整的表面上,并最終導致無法控制的生長。此外,枝晶尖端的電流強度明顯高于其他區域,表明枝晶將進一步生長(圖3g)。相比之下,鋅尖端的電場和電流密度被BA添加劑屏蔽,使鋅能夠均勻沉積(圖3h和3i)。基于上述結果,BA在增強耐腐蝕性和抑制HER副反應中的作用如圖3j所示。
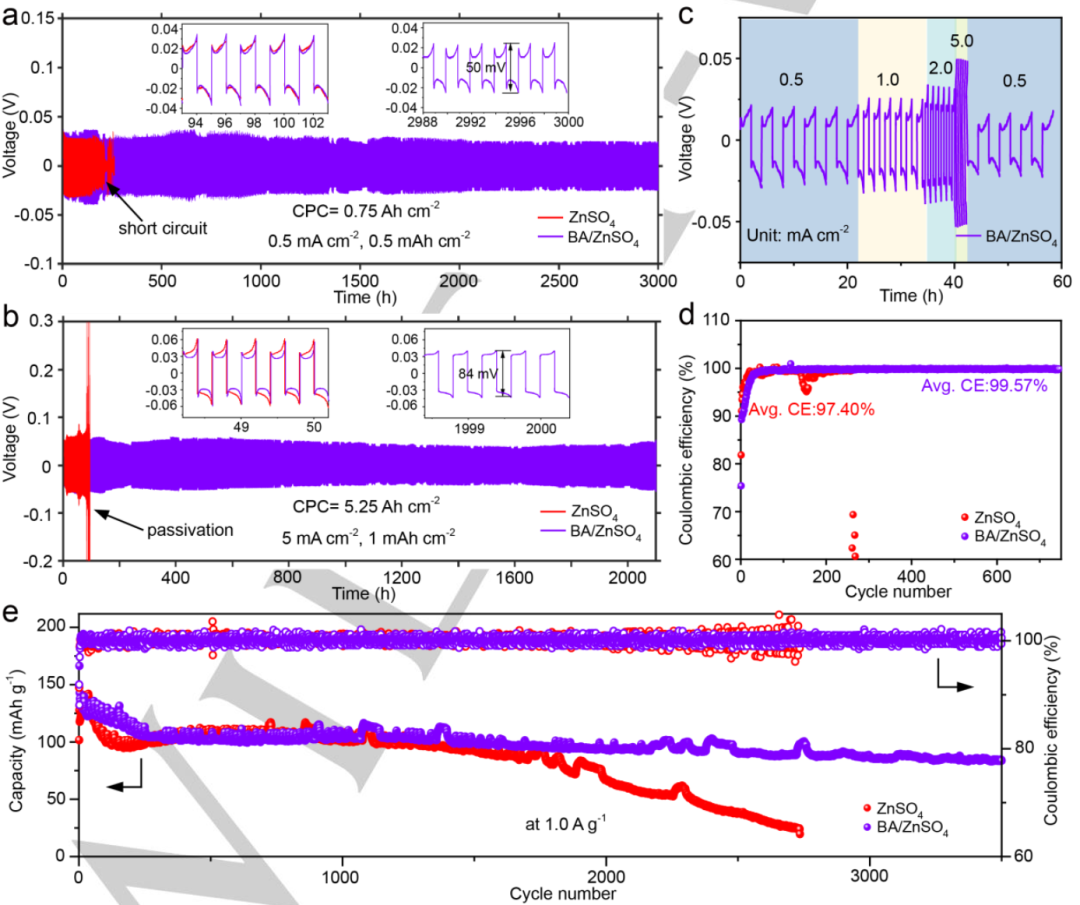
圖4.在a)0.5 mA cm-2、0.5 mAh cm-2和b)5 mA cm-2、1 mAh cm-2下添加或不添加BA的Zn//Zn對稱電池循環性能。c)使用BA/ZnSO4電解質的Zn//Zn對稱電池倍率性能。d)不同電解液中,1 mA cm-2@0.5 mAh cm-2下Zn電鍍/剝離的CE。e)具有兩種電解質的Zn//MnO2電池在1.0 A g-1下的循環穩定性和效率。@Wiley
圖4a顯示,添加BA添加劑后,Zn//Zn對稱電池在0.5 mA cm-2@0.5 mAh cm-2條件下可以穩定循環超過3000小時,而沒有BA添加劑的電池在210小時后會短路。即使在更高的電流密度(5 mA cm-2@1 mAh cm-2)(1.9% 的放電深度,DOD)下循環,使用BA/ZnSO4電解質的對稱電池也能實現2100小時的超長循環壽命(圖4b),幾乎是沒有BA添加劑(約100小時)的21倍。圖4c顯示,使用BA添加劑后,Zn//Zn對稱電池在所有電流密度下都表現出穩定的電壓曲線。
圖4d顯示,在沒有BA添加劑的Zn//Cu電池前268個循環中,平均CE為97.4%。但CE值在隨后的循環中波動。相比之下,具有BA添加劑的Zn//Cu電池CE明顯提高,平均值為99.57%,這是因為BA取代了溶劑化鞘中活性較低的水分子。圖4e顯示,添加BA添加劑的電池可以穩定循環3500次,進一步證明BA可以顯著提高鋅電池的循環穩定性。
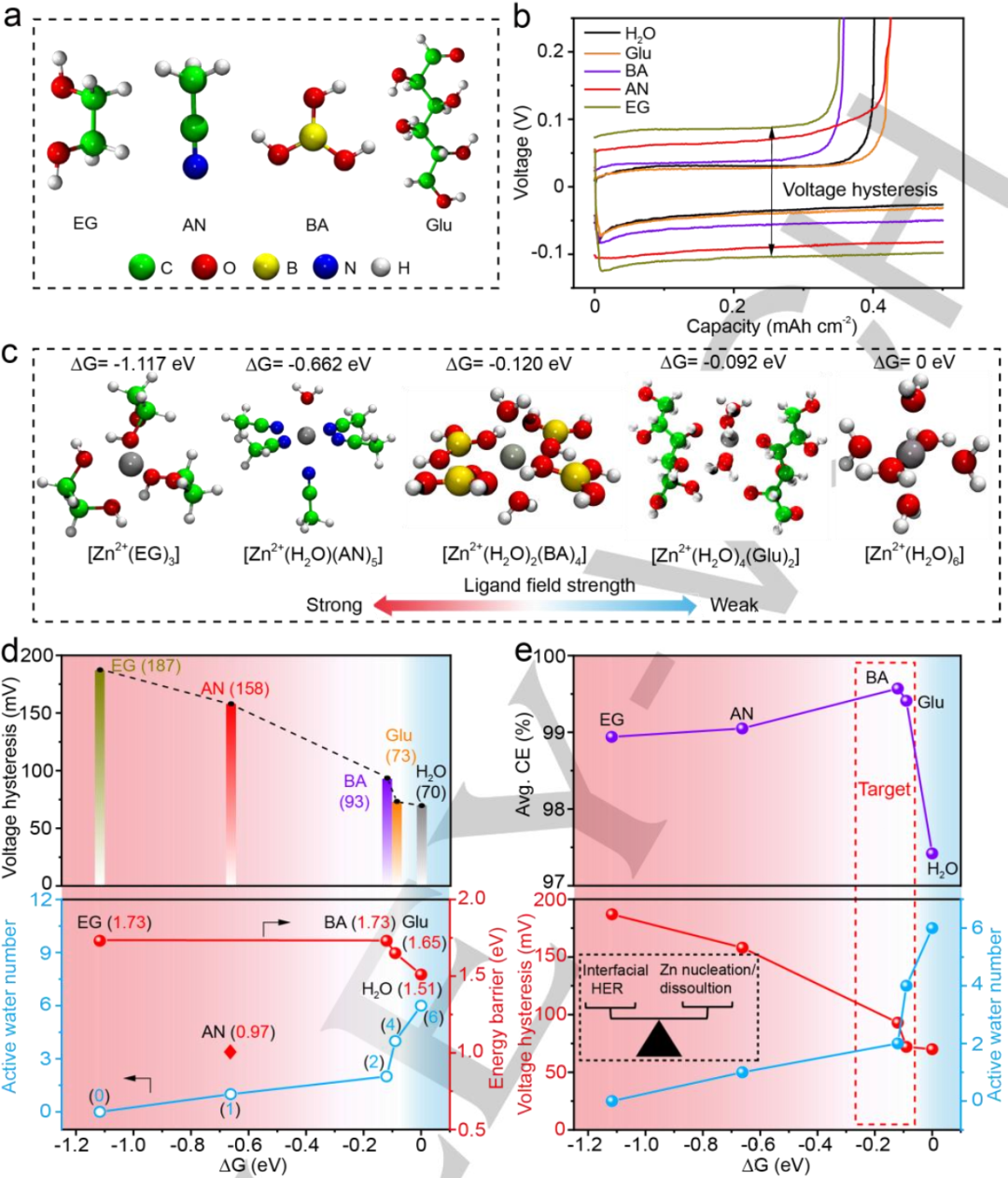
圖5.?a)不同添加劑的結構。b)引入不同添加劑后第一個循環的電壓曲線。c)不同添加劑存在下優化的Zn2+溶劑化鞘和相應的配體場強順序。d)溶劑化鞘ΔG、Zn成核/溶解動力學(頂部)和界面HER(底部)之間的相關性。e)ΔG、電壓極化、溶劑化鞘中的活性水數量和平均CE之間的相關性。@Wiley
接下來,利用DFT計算系統地評估了溶劑化鞘的熱力學穩定性和水解離動力學。除了BA分子外,還考慮了文獻中常用的添加劑EG、AN、和Glu分子,如圖5a所示。圖5b顯示了添加了EG、AN、BA、Glu和H2O的Zn//Cu半電池電壓極化。其中EG、AN、BA、Glu和H2O的電壓極化分別為187、158、93、73和70 mV,說明添加劑的引入顯著提高了Zn成核/溶解的能壘。
為了進一步探究各種添加劑與Zn2+在Zn成核/溶解過程中的相互作用,計算了溶劑化結構的吉布斯自由能(ΔG)。在不同添加劑作用下,最穩定的溶劑化結構如圖5c所示,其中EG、AN和Glu的電解質添加劑更傾向于與Zn2+配位,分別形成穩定的[Zn2+(EG)3]、[Zn2+(H2O)(AN)5]和[Zn2+(H2O)4(Glu)2]溶劑化鞘。溶劑化鞘對應的ΔG值順序為EG>AN>BA>Glu>H2O。
此外,Zn2+核與周圍配體分子之間的溶劑化鞘基本可以用配體場理論來描述。具有雙齒配體的EG表現出最強的配體場,而具有弱?鍵的水表現出最弱的配體場。其中BA配體由于π*反饋鍵的形成,具有適度的配體場。因此,形成的穩定溶劑化鞘配體場強度順序與ΔG的趨勢很好地吻合。
為了將計算得到的溶劑化鞘ΔG與Zn成核/溶解動力學和HER抑制能力聯系起來,圖5d繪制了溶劑化鞘的活性水數量、電壓極化與ΔG之間的關系。ΔG與[Zn2+(EG)3],[Zn2+(H2O)(AN)5],[Zn2+(H2O)2(BA)4],[Zn2+(H2O)4(Glu)2]和[Zn2+(H2O)6]的溶劑化鞘初始電壓極化呈正相關。
此外,隨著配體場的增大,ΔG減小,相應的電壓極化變大。其中[Zn2+(H2O)6]由于初始極化電壓較小,具有最佳的Zn2+成核溶解動力學。但是,第一溶劑化殼層中水分子的充分占據會大大加速Zn表面的HER動力學。因此,接下來探討了電解質添加劑對界面HER動力學的影響。如圖5d(下)所示,較強的配體場對應較低的ΔG,而在原始的[Zn2+(H2O)6]溶劑化鞘中,更多的水分子被配體添加劑取代。
圖5e顯示,ΔG不僅與初始電壓極化具有很強的相關性,而且與活性水數量和平均CE呈火山狀關系,這意味著理想的溶劑化鞘不應具有太強或太弱的 ΔG(以及配體場強),從而實現出色的Zn成核/溶解動力學并抑制HER副反應。
05 ? 成果啟示
該工作證明了BA配體與Zn2+核具有適度的配體場相互作用,從而實現了良好的Zn成核/溶解動力學并抑制了水的解離。XAFS和NMR測試表明,BA僅通過配體交換部分取代Zn2+配位層中的水分子,形成了一個穩定的缺水溶劑化鞘。添加BA的Zn//Zn對稱電池在5 mA cm-2下能夠實現5.25 Ah cm-2的累積電鍍容量和84 mV的超低電壓極化。
BA和之前報道的電解質添加劑BA、AN、EG、Glu的配體場強度相比,Zn2+溶劑化鞘的相對自由能與Zn成核/溶解動力學和HER抑制能力有很強的相關性,表現出典型的火山行為。配體場強與副反應動力學之間的相關性為合理設計快速和可逆的鋅電池提供了新的思路。
審核編輯:劉清