【研究背景】
鋰離子電池(LIBs)的能量密度和電壓較高,是最有前途的存儲系統之一。對于LIBs綜合性能的進一步提升,鋰金屬負極的使用是必然趨勢。然而,鋰金屬化學活性較高,在傳統的有機電解液體系中更容易引發安全風險。因此,全固態電池(ASSBs)應運而生。然而,目前缺少商業化的高導電性固態電解質,同時無枝晶的鋰金屬可逆循環也是一大問題。
關于高性能固態電解質的研發目前已有一定進展,如陽離子取代的Li7La3Zr2O (LLZO)衍生化合物,離子電導率高,對鋰金屬的動力學穩定性,可以在空氣氣氛中工作。然而,其容易開裂,機械性能較差,對鋰枝晶的形成無法形成彈性緩釋。硫化物雖然對水敏感且不耐氧化,然而其離子電導率較高,且機械性能好。富鋰三元磷化物目前也被認為是有前景的固態電解質,P3-比硫化物陰離子具有更大的極化率。
盡管上述固態電解質各有千秋,卻有一個共性問題—對金屬鋰本征熱力學不穩定。因此,開發兼具高離子電導和對金屬鋰穩定性高的固態電解質,必須綜合考慮結構和熱力學特性以及合成方法。
【成果簡介】
作者利用高通量晶體結構預測輔助簡單的高能球磨技術,將Li3P和Li2S全無序化以制備固態電解質,并通過多種表征分析方法證明其不僅具備高離子電導率,對于金屬鋰還具備本征的熱力學穩定性。該工作為高性能固態電解質的開發提供了理論指導和方法借鑒。
【研究亮點】
(1)利用高通量晶體結構預測技術,首次預測并發現Li-P-S三元體系具備高性能固態電解質的潛質,并繪制出相圖。
(2)利用簡單的固相球磨法使Li3P和Li2S完全無序化,制得的固態電解質具備高鋰離子電導率和對金屬鋰的本征穩定性,實現了前期預測。
【圖文導讀】
首先對體系進行理論分析,如圖1所示,所有具備低形成能的新相均位于相圖的邊界處。作者通過結構預測發現了新的亞穩態Li2S?P2S5三元化合物,即Li5P3S10和Li5PS5,分別位于Li?P?S上方31.2和54.4 meV/原子處。
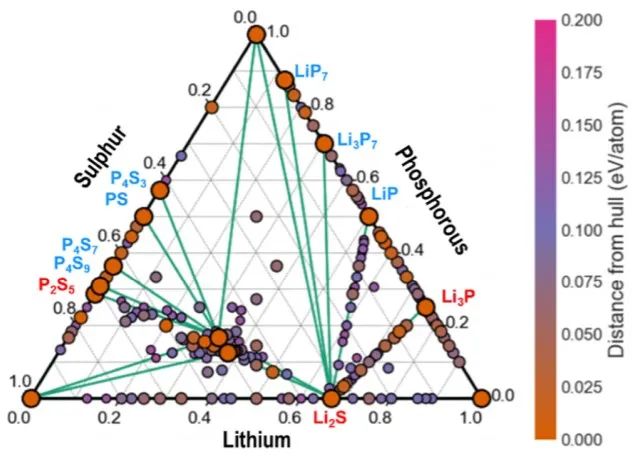
圖1 Li?P?S三元相圖及用DFT/ PBE隨機結構分布. 頂點為Li/P/S純相,邊為二元相。
經過進一步分析,源自基礎材料(Li2S和Li3P)的一些新結構在預測得到的三元基態結構中比較常見,即Li8P2S和Li5PS,分別是Li2S/Li3P的1:2和1:1的混合物(圖2)。這表明兩個相圖中的頂點組分可能形成共生相。如圖3所示,固溶體的衍射譜圖都可以用一個基于Li2S反螢石晶體結構的模型進行對比測試,該晶體結構由硫化物陰離子的立方密堆積(ccp)組成。
由于S2?被較大的P3?取代,大量的Li3P使晶胞參數逐漸增加,當x=0.33, 0.50, 0.67 和0.75時,晶胞參數分別為5.83(8), 5.90(6), 5.96(3) 和5.98(6) ?。盡管在環境壓力和溫度下不同時的晶體結構不同,但晶格參數的變化依然遵循維加德定律。
?
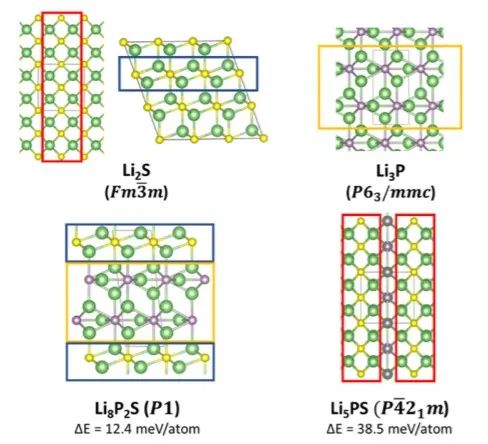
圖2 基態結構Li2S和Li3P以及新預測的Li8P2S和Li5PS三元相。
?
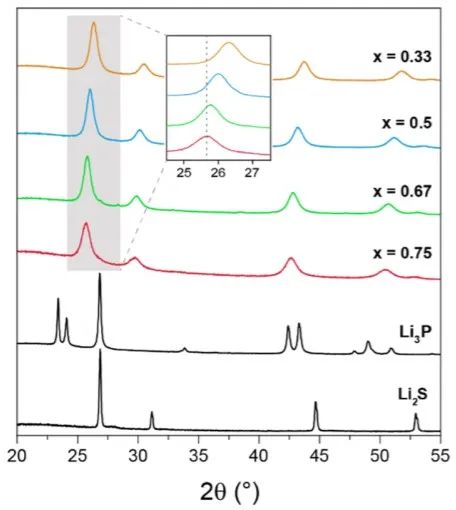
圖3 xLi3P-(1-x)Li2S的XRD測試結果。 測得的XRD譜圖與兩種可能的晶體結構模型一致(圖4a),在模型1的反螢石型衍生結構中,陰離子位置(Wyckoff 4a位點)具有P3?/S2?占位障礙,而額外的Li+則可被容納在八面體空位中。模型2中,受限于電中性原則,額外的Li位點是不存在的。
?
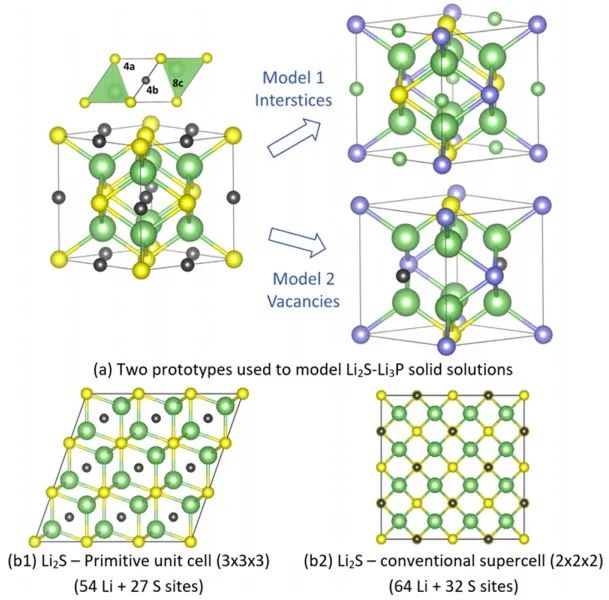
圖4 (a) Li2S?Li3P固溶相的兩種可能的結構模型;(b) 采用Li2S超胞作為缺陷構型起點的DFT計算結果。 圖5a-b顯示了x=0.5樣品XRD和中子衍射共擬合結果。如圖(a)所示,使用模型2對中子衍射數據精細擬合效果不佳,而基于模型1的擬合則成功地對中子衍射和同步輻射XRD譜圖進行了建模。此外,XRD和NPD模式中觀察到的反射較廣,原則上這可以歸因于高能球磨合成條件下產生的小晶粒尺寸或高應變。
?
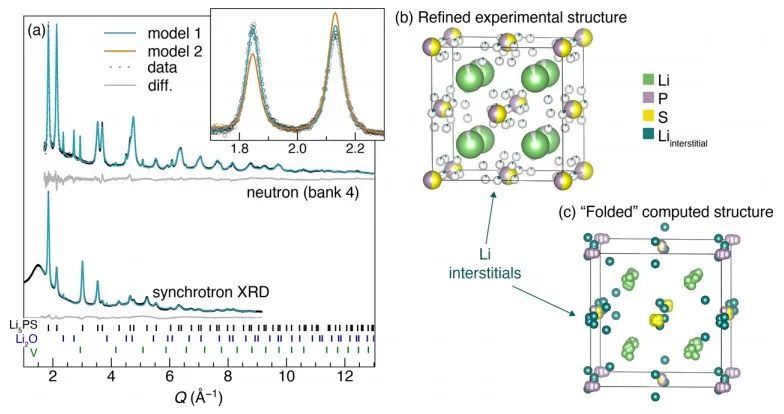
圖5 對x=0.5樣品(Li5PS)的同步輻射XRD和中子粉末衍射測試分析結果。(a)對XRD和中子衍射的Rietveld精修擬合結果,結構模型如圖(b)所示,對應于模型1. (c) DFT弛豫計算,體系結構為x=0.5,其為折疊成單一的常規反螢石結構。 采用31P和6/7Li固態MAS NMR研究了固溶體的局域結構(圖6)。7Li譜顯示,對于純的二元組分,2.2 ppm為Li2S信號,4.5和7.1 ppm則對應Li3P信號,確切的位移是由Li2S和Li3P的比值決定的。
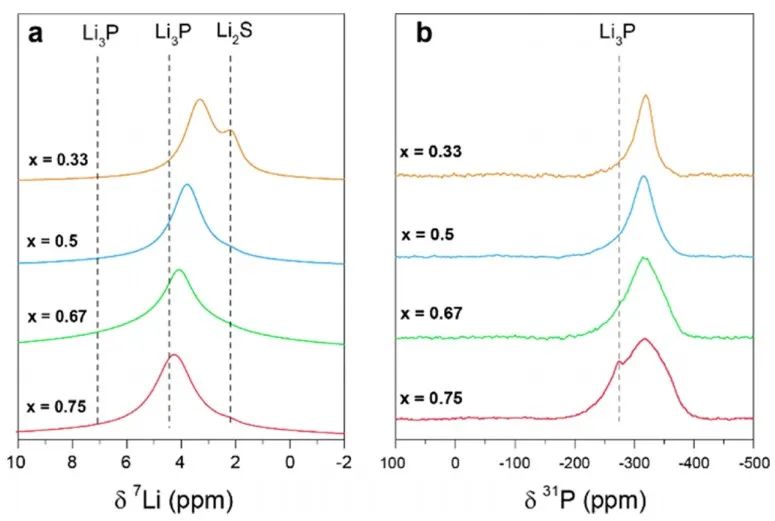
圖6 室溫下xLi3P-(1-x)Li2S的(a)7Li和(b)31P的MAS NMR測試結果。 在進行VT測量之前,作者首先評估了樣品在高溫下的穩定性。在光譜儀內加熱樣品到125°C, 7Li NMR譜線寬度減小(圖7)。當樣品在該溫度下保持約1h時,線寬繼續減小,這可能是樣品和觀測到的原子核分別在ns和μs時間尺度上的燒結所致。
相關時間與弛豫時間成正比,在不相關的三維運動中,阿倫尼烏斯行為十分顯著,由此可計算得到體系活化能(圖8)。對于固相體系,弛豫行為還表現出運動維數效應和相關效應,這會導致弛豫曲線的高、低溫側的表觀活化能不同。
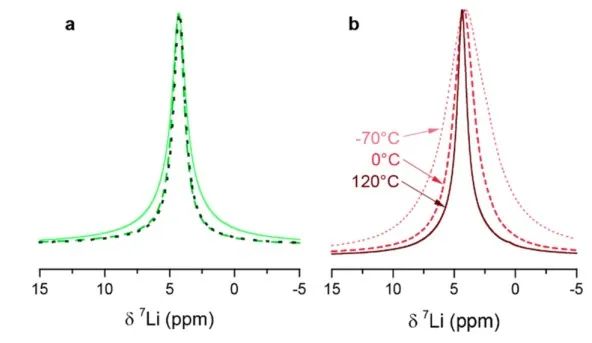
圖7 (a) x=0.67樣品直接球磨后的室溫NMR譜(實線),在125℃退火后的1小時(虛線)、2小時(虛線). (b) x=0.75樣品的溫度相關性NMR譜。
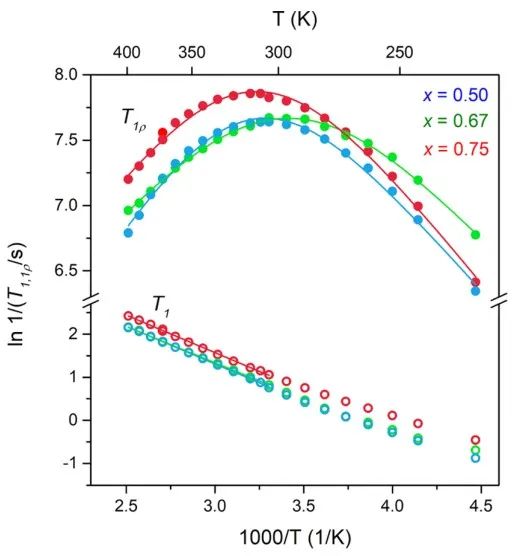
圖8 xLi3P-(1?x)Li2S在-50-125℃下的7Li信號弛豫時間。 作者使用VT-EIS進一步探測鋰離子遷移率,四個樣品的室溫阻抗如圖9所示。所有樣品的EIS圖中都僅觀察到一個半圓,因此無法區分晶粒(體積)和晶界的貢獻。
對于x=0.5的樣品,溫度從100°C到RT和RT到?50°C的代表性VT-EIS圖分別如圖10a-b所示,所有樣品的電導率都與溫度呈線性關系。Arrhenius圖給出的活化能Ea在0.2~0.15 eV之間,這與核磁共振弛豫測量得到的值一致,x=0.67樣品的Ea最低。
?
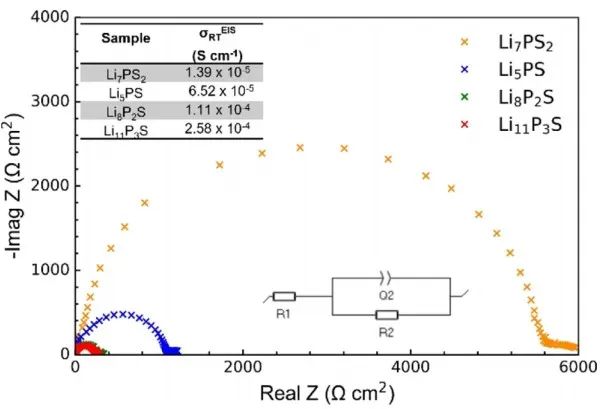
圖9 xLi3P-(1-x)Li2S不同組分的Li|SE|Li對稱電池的室溫EIS圖。
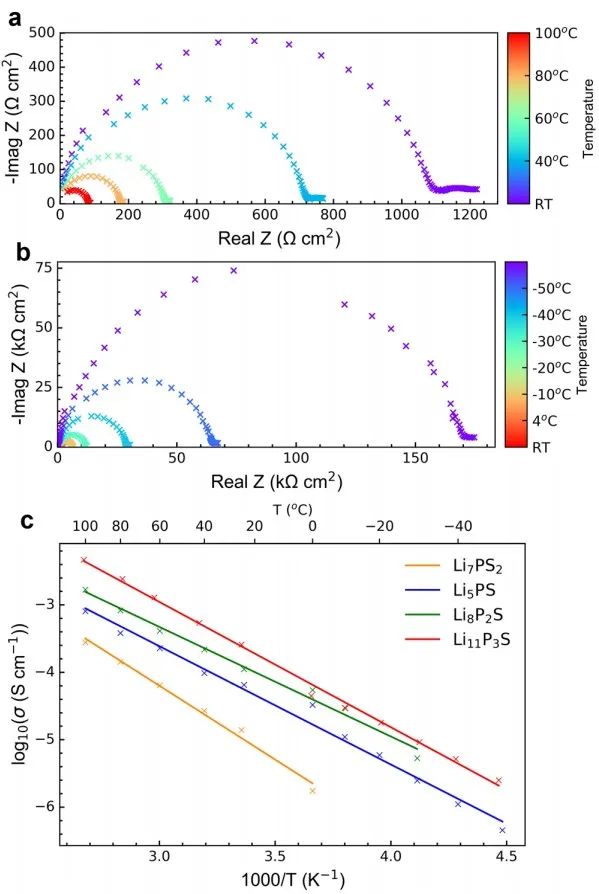
圖10 x = 0.5樣品在(a) 100°C至RT和(b) RT至?50°C時,Li|SE|Li對稱電池的EIS圖;(c) EIS-Arrhenius曲線顯示了不同xLi3P-(1?x)Li2S組成的總離子電導率與溫度的關系。 將通過DFT/GIPAW計算得到的x=0.67時7Li和31P NMR位移與實測結果進行了比較(圖11),通過在298 K處的玻爾茲曼常數作為比例因子得到該測試結果。
基于以上結果,作者提出了一種基于反螢石的結構模型,其中S2-/P3-混合陰離子晶格為滿占據狀態,Li+(部分)占據四面體和八面體位點,空位則允許Li+躍遷。對所有成分進行類似的CE分析,得出Li2S?Li3P二元相圖(圖12)。
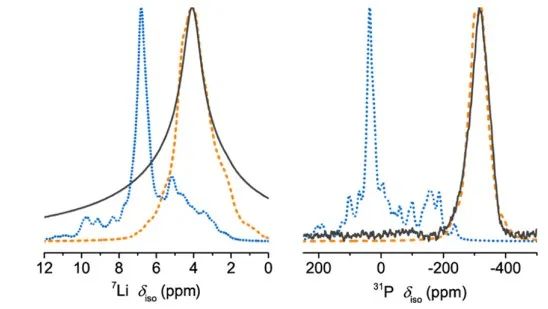
圖11 Li8P2S三元相的預測7Li和31P NMR譜。
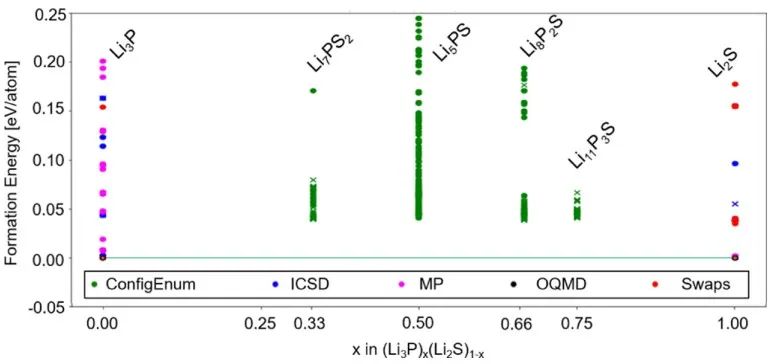
圖12 Li3P?Li2S二元組分的預測結果(0 K)。
為了進一步驗證預測的Li?P?S三元結構(圖13),作者比較了計算的以及實測的7Li和31P NMR譜。預測的重心漂移分布隨P含量的增加而變化愈發不明顯。然而,在實測光譜中發現,這可能是因為計算沒有考慮Li亞晶格的遷移所致。 如圖14,作者還比較了離子電導率,并計算了相應的活化能(Ea),AIMD模擬結果與Li?P?S體系中已知的高電導率三元化合物進行了比較,當P含量越高時,由NMR和EIS得到的活化能越高。
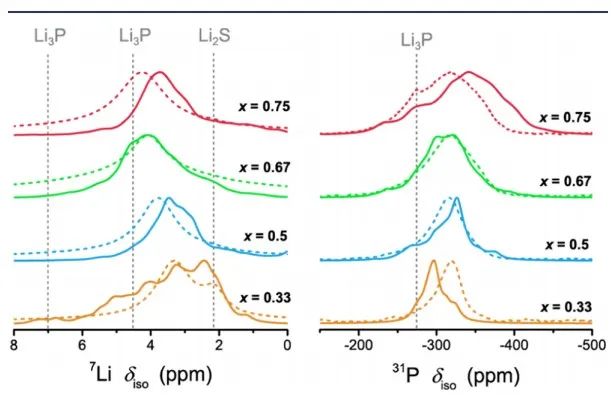
圖13 Li-P-S的7Li和31P NMR譜的預測結果(實線)和實測結果(虛線)。
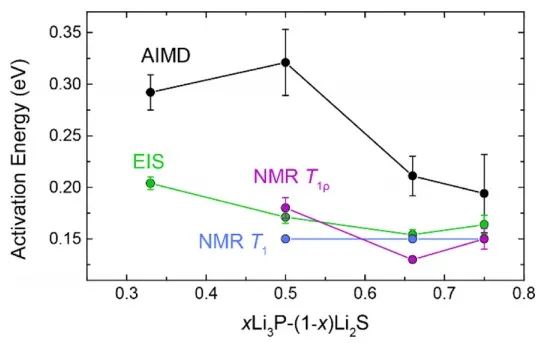
圖14 預測和實驗實測的 Li2S?Li3P活化能(Ea)。
【總結和展望】
本文報道了一種新的xLi3P?(1?x)Li2S固溶體的合成,以及結構解析和離子電導率測量,并以量子化學計算為支持。兩種二元化合物高能球磨后,在0.39≤x≤0.75范圍內形成反螢石結構類型的固溶體,最大的特點在于其整體為無序的陰離子晶格構型,四面體和八面體空洞被移動的Li+填充。這種材料具備高導電性和對金屬鋰本征的穩定性,因此可以被認為是潛在理想的固態電解質。該工作為開發高性能全固態電池提供了新的思路和見解。
審核編輯:劉清