01 ? 導讀
在傳統(tǒng)可充電鋰離子電池(LIB)中,以碳酸亞乙酯(EC)為主的Li+初級溶劑化鞘(PSS)在Gr上能夠形成獨特的固體電解質(zhì)界面(SEI),抑制溶劑的共嵌入,并避免Gr的結(jié)構(gòu)坍塌。為了提高電池安全性,在EC基電解質(zhì)中引入有機磷酸鹽作為共溶劑,可以增加電解液的阻燃性。
然而,大多數(shù)有機磷酸鹽表現(xiàn)出高的極性和強的給電子能力,可以與Li+配位。因此,磷酸鹽分子進入PSS并部分取代EC分子(圖1a)。通過這種方式,磷酸鹽參與了初級SEI的形成,并破壞了原來的SEI。此外,配位磷酸鹽傾向于與Li+共嵌入Gr晶格中。插層分子在低電位下的分解會使Gr顆粒破裂,并導致性能迅速下降。因此,在不干擾電極材料儲鋰性能的情況下構(gòu)建阻燃電解質(zhì)仍然具有挑戰(zhàn)性。
02 ? 成果背景
近日,JACS上發(fā)表了一篇題為“Noncoordinating Flame-Retardant Functional Electrolyte Solvents for Rechargeable Lithium-Ion Batteries”的文章,該文章提出了一種非配位阻燃共溶劑,能夠提高電池安全性,同時避免對儲鋰過程的干擾。選用具有良好理化性能的高效阻燃劑六氟環(huán)三磷腈作為共溶劑制備功能電解質(zhì)。
給電子能力低的非極性磷腈分子不能與Li+配位,因此被排除在初級溶劑化鞘外。在基于石墨負極的鋰離子電池中,磷腈分子在充電過程中不會與鋰離子共插進入石墨晶格中,有助于保持負極結(jié)構(gòu)和界面的完整性,實現(xiàn)穩(wěn)定循環(huán)。
03 ? 關(guān)鍵創(chuàng)新
(1)六氟環(huán)三磷腈(HFP)作為一種非配位的阻燃共溶劑,不參與構(gòu)建Li+溶劑化鞘,因此不會破壞原有的SEI結(jié)構(gòu)。
(2)HFP不與Li+共插入石墨層間,因此避免了石墨負極結(jié)構(gòu)和界面的破壞。
(3)HFP的高阻燃特性極大提高了電池安全性。
04 ? 核心內(nèi)容解讀
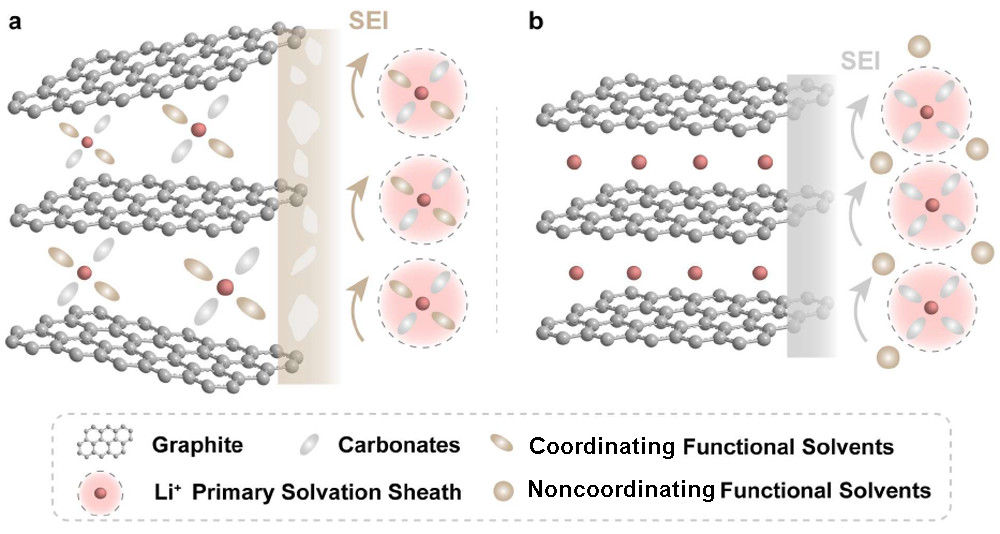
圖1.?在(a)具有配位共溶劑的電解質(zhì)和(b)具有非配位共溶劑的電解質(zhì)中,Li+嵌入Gr的示意圖。
本工作提出了一種新的非配位共溶劑——六氟環(huán)三磷腈(HCP),它可以提高電解質(zhì)阻燃性,但對Li+溶劑化結(jié)構(gòu)的影響最小(圖1b)。HCP是一種全氟六元芳族磷腈分子,作為阻燃共溶劑,被引入傳統(tǒng)的碳酸鹽電解質(zhì)E1(0.8 M LiPF6?in?EC/碳酸二乙酯(DEC),1:1.5 m/m),獲得功能性電解質(zhì)E2(0.8 M LiPF6?in EC/DEC/HCP,11 mm)。通過將經(jīng)典的磷酸鹽TMP加入E1來制備電解質(zhì)E3(0.8 M LiPF6?in EC/DEC/TMP,11 mm),并作為對照組。E1和E3可點燃并劇烈燃燒。相反,E2不能被點燃。
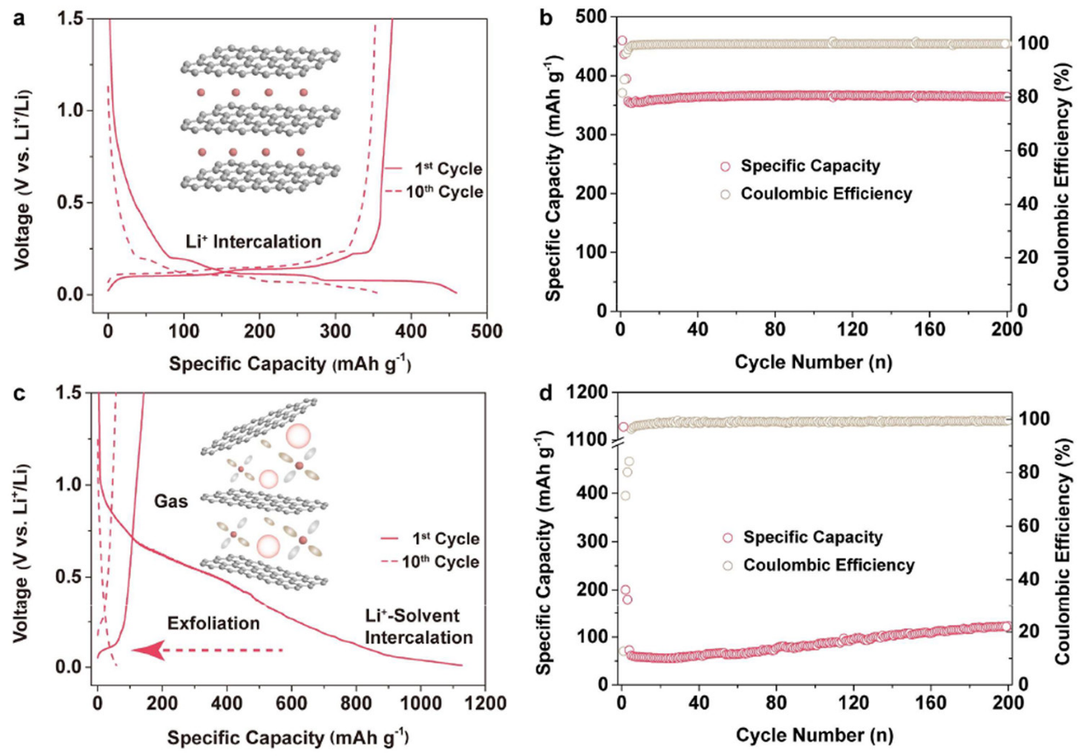
圖2.?使用(a, b)E2和(c, d)E3電解質(zhì)的Li||Gr電池(a, c)恒流充放電曲線和(b, d)循環(huán)性能。
作為鋰離子電池最常用的負極材料,Gr對電解質(zhì)和SEI有著嚴格的要求。為了研究上述電解質(zhì)的電化學相容性,對Li||Gr電池進行了測試。在初始放電過程中,Li|E1|Gr和Li|E2|Gr電池的比容量均約為460 mAh g-1,具有三個電壓平臺,分別對應(yīng)于SEI形成和Li+嵌入Gr(圖2a)。相比之下,Li|E3|Gr電池的比容量大于1000 mAh g-1,電壓曲線表明發(fā)生了大量溶劑共嵌入和分解(圖2c)。
Li|E1|Gr和Li|E2|Gr表現(xiàn)出穩(wěn)定的循環(huán)性能,并在200次循環(huán)后,保持約100%的初始充電容量(圖2b),而Li|E3|Gr容量迅速下降(圖2d)。顯然,將HCP引入碳酸鹽電解質(zhì)不會影響與Gr的相容性。
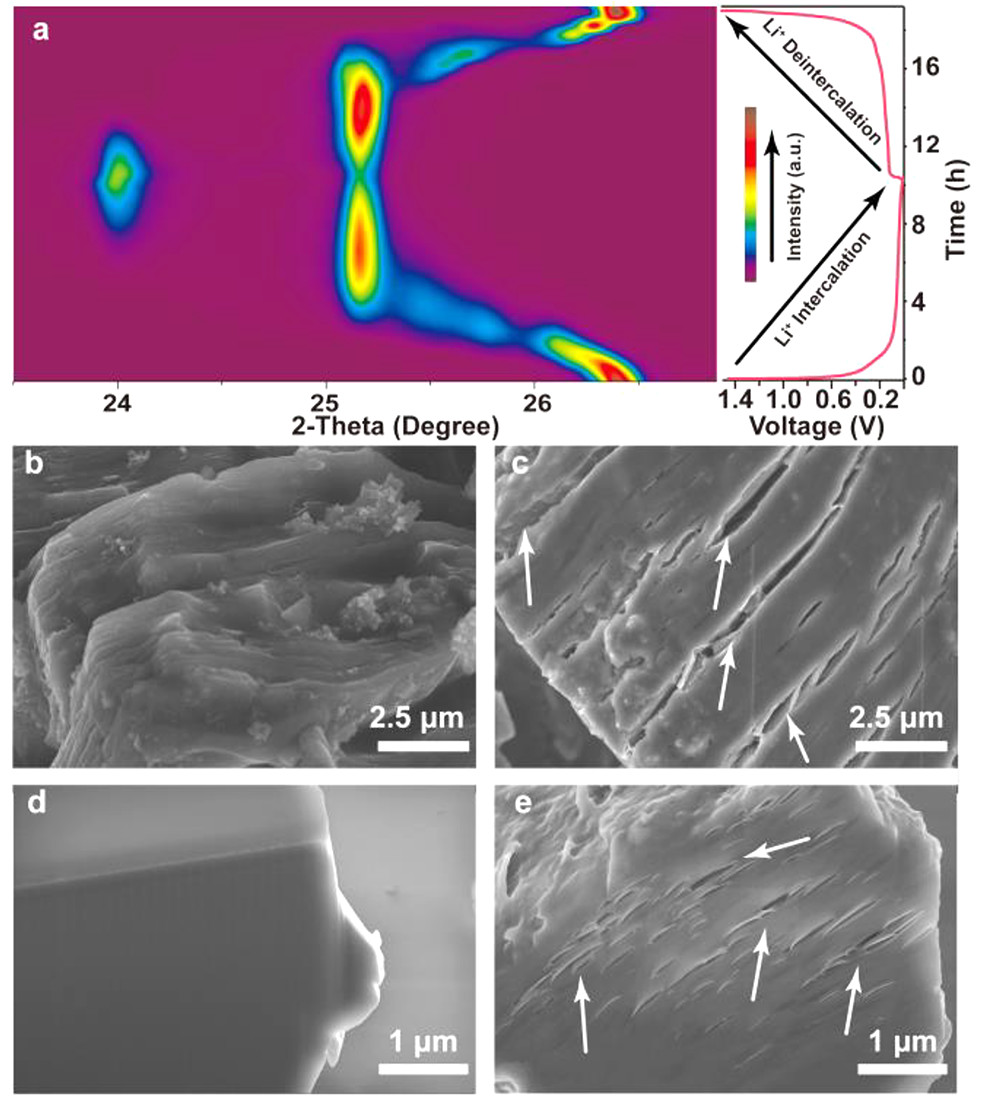
圖3.?Li|E2|Gr電池(a)第一次充放電循環(huán)(0.1C, 0.01-1.5 V)的原位XRD等高線圖。在(左)E2和(右)E3電解質(zhì)中第一次循環(huán)后,Gr的(b, c)SEM圖和(d, e)FIB-SEM圖。@ACS
接下來,研究了在不同荷電狀態(tài)下Gr的X射線衍射(XRD)譜圖。Li|E2|Gr電池充放電過程的原位XRD譜圖(圖3a)顯示,Gr的(002)峰位置在Li+插層過程中由26.4°變?yōu)?3.7°,放電過程中向相反方向?qū)ΨQ移動,表明Li+插層具有高的可逆性。SEM圖像顯示,在E3中循環(huán)后的Gr顆粒明顯膨脹,表面(圖3c)和內(nèi)部(圖3e)均有明顯裂紋。
相反,E2(圖3b,d)中循環(huán)的Gr顆粒保持良好,沒有可見裂紋。這些明顯的裂紋和Gr在E3中的巨大徑向膨脹(約10倍)可能是由于共插的溶劑分子分解導致層狀結(jié)構(gòu)剝落。
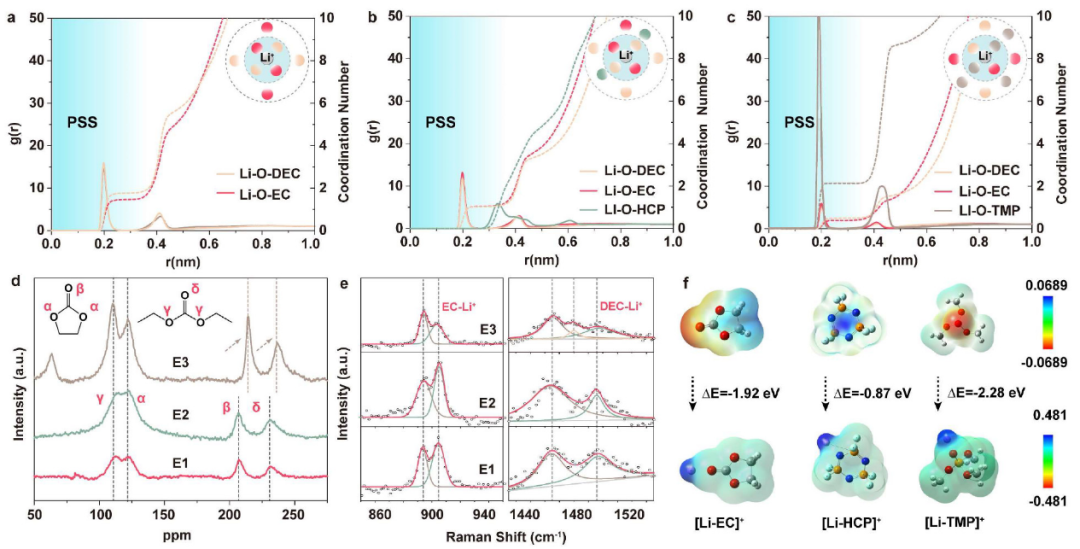
圖4.?(a-c)不同電解質(zhì)的Li-O徑向分布函數(shù)。所附原理圖顯示了相應(yīng)的溶劑化結(jié)構(gòu)。不同電解質(zhì)的(d)17O核磁共振譜和(e)拉曼光譜。(f)溶劑和溶劑化結(jié)構(gòu)的靜電勢分布。@ACS
接下來,通過分子動力學(MD)模擬來說明Li+在不同電解質(zhì)中的溶劑化結(jié)構(gòu)(圖4a-c)。首先,對溶劑化層中與Li+相互作用的溶劑分子進行計數(shù)和平均。如圖4c所示,E3的Li+?PSS中存在TMP,平均配位數(shù)為2.13,遠遠大于EC和DEC的配位數(shù)(分別為0.39和0.49)。相比之下,HCP幾乎沒有參與PSS(r<3 ?),E2中的平均配位數(shù)接近于0(圖4b)。與E1的情況一樣(圖4a),由于HCP的非配位性,EC和DEC在E2的Li+?PSS中仍然占主導地位。
采用密度泛函理論(DFT)模擬計算溶劑和溶劑化結(jié)構(gòu)的形成能和靜電勢(圖4f)。相對于HCP, TMP和EC具有更強的表面負電荷,表現(xiàn)出更強的給電子能力和Li+配位能力。[Li+-溶劑]形成能可定義為
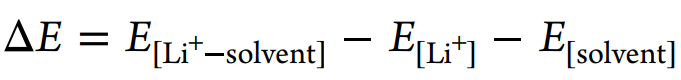
ΔE[Li+-EC](?1.92 eV)比ΔE[Li+-HCP](?0.87 eV)大得多,但小于ΔE[Li+TMP](?2.28 eV),說明這三種溶劑的配位能力為TMP>EC?HCP, HCP不能在熱力學上取代EC。
通過17O核磁共振光譜、拉曼光譜和電噴霧質(zhì)譜(ESI-MS),進一步研究了電解質(zhì)的溶劑化結(jié)構(gòu)。如圖4d所示,EC和DEC中有兩組不等效的17O核,羰基17O(β和δ)位于200和250 ppm之間,醚17O(α和γ)位于75和150 ppm之間。E3中羰基17O核經(jīng)歷了下場位移,脫屏蔽效應(yīng)增強,這可能與TMP參與Li+?PSS以及Li+與碳酸鹽溶劑之間離子偶極相互作用減弱有關(guān)。
相反,E2中沒有發(fā)現(xiàn)位移,說明HCP不影響[Li+-碳酸鹽]的溶劑化結(jié)構(gòu)。905 cm-1處的拉曼峰(圖4e)代表[Li+-EC]鍵。相對于E1, E3中905 cm-1處的峰強度明顯降低,而E2中無明顯變化。在1486 cm-1處的[Li+-DEC]峰也可以觀察到類似的結(jié)果。這表明HCP對碳酸鹽的溶劑化狀態(tài)影響較弱,證實了其非配位性。
05 ? 成果啟示
本工作提出了一種非配位的磷腈共溶劑,以提高電解質(zhì)的阻燃性,同時避免干擾LIBs中的電荷轉(zhuǎn)移和存儲過程。與傳統(tǒng)的有機磷酸鹽不同,非配位HCP分子不參與Li+?PSS的形成,也不與Li+共插到負極結(jié)構(gòu)中,因此不會導致負極-電解質(zhì)界面退化或SEI不穩(wěn)定。基于該新型電解質(zhì)的LIB具有良好的循環(huán)性能。該新型非配位阻燃共溶劑與其他正極穩(wěn)定劑結(jié)合,有望協(xié)同提高可充電電池的存儲和安全性能,在未來的電化學系統(tǒng)中具有廣闊的應(yīng)用前景。
審核編輯:劉清