鋰離子電池正極材料LiNiVO4的容量衰減原因分析
摘要:通過流變相反應(yīng)法在600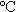 合成了純相的鋰離子電池正極材料LiNiVO4。采用非現(xiàn)場X射線衍射(ex-situ XRD)、循環(huán)伏安(CV)、電化學(xué)阻抗譜(EIS)和掃描電子顯微鏡(SEM)研究了其容量衰減的原因。結(jié)果表明,LiNiVO4在循環(huán)過程中的結(jié)構(gòu)是可逆的;但LiNiVO4與電解質(zhì)溶液較差的相容性及高電壓下電解液的分解是其容量衰減的主要原因。
合成了純相的鋰離子電池正極材料LiNiVO4。采用非現(xiàn)場X射線衍射(ex-situ XRD)、循環(huán)伏安(CV)、電化學(xué)阻抗譜(EIS)和掃描電子顯微鏡(SEM)研究了其容量衰減的原因。結(jié)果表明,LiNiVO4在循環(huán)過程中的結(jié)構(gòu)是可逆的;但LiNiVO4與電解質(zhì)溶液較差的相容性及高電壓下電解液的分解是其容量衰減的主要原因。
關(guān)鍵詞:鋰離子電池;正極材料;LiNiVO4;容量衰減
LiNiVO4是一個具有反尖晶石結(jié)構(gòu)的化合物,它是由Fey G T K等首次提出來的[1]。在LiNiVO4反尖晶石結(jié)構(gòu)中,Li和Ni原子被平均隨機分布在八面體間隙中,而V原子則占據(jù)四面體間隙位置,形成Vtetra(LiNi)octaO4。反尖晶石LiNiVO4在XRD圖譜中最明顯的特征是(111)衍射峰明顯變?nèi)酰?220)衍射峰則明顯增強。Liu R S等利用Rietveld法分析精修了LiNiVO4的晶體學(xué)數(shù)據(jù)[2],除了表明LiNiVO4為反尖晶石結(jié)構(gòu)外,其認為仍有約8%的V原子占據(jù)八面體位置,也有約8%的Ni原子占據(jù)八面體位置。LiNiVO4既可作為鋰離子電池的正極材料也可作為作為鋰離子電池的負極極材料。當其作為鋰離子電池正極材料時,其具有很高的充放電平臺(4.8V)[1,2],在提高鋰離子電池比能量方面具有潛在的應(yīng)用價值。
然而,與尖晶石型的正極材料相比,鋰離子LiNiVO4中的傳輸相對困難[3],這導(dǎo)致了其放電容量較低。但大量的研究表明[4-8],LiNiVO4作為鋰離子電池正極材料時的最大問題是其放電容量衰減較快。在本文中,我們采用流變相反應(yīng)法合成LiNiVO4,初步探討了LiNiVO4的容量衰減原因,旨在為改善LiNiVO4的電化學(xué)性能提供思路。
1 實驗部分
1.1 LiNiVO4材料的合成
將LiOH H2O、Ni(CH3COO)2
H2O、Ni(CH3COO)2 4H2O、NH4VO3和C2H2O4
4H2O、NH4VO3和C2H2O4 2H2O按化學(xué)計量比1:1:1:2的比例稱取后充分研磨,然后加入適量的二次蒸餾水調(diào)成均勻的流變態(tài)混合物。將流變態(tài)混合物轉(zhuǎn)移到流變相反應(yīng)釜內(nèi)并于90
2H2O按化學(xué)計量比1:1:1:2的比例稱取后充分研磨,然后加入適量的二次蒸餾水調(diào)成均勻的流變態(tài)混合物。將流變態(tài)混合物轉(zhuǎn)移到流變相反應(yīng)釜內(nèi)并于90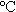 下保溫5h,然后取出反應(yīng)物于120
下保溫5h,然后取出反應(yīng)物于120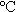 烘干、研磨制得棕紅色LiNiVO4前驅(qū)體。將前驅(qū)體在600
烘干、研磨制得棕紅色LiNiVO4前驅(qū)體。將前驅(qū)體在600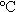 空氣氛中加熱10h后,冷卻至室溫取出研磨制得LiNiVO4。
空氣氛中加熱10h后,冷卻至室溫取出研磨制得LiNiVO4。
1.2 LiNiVO4材料的表征
采用X射線粉末衍射儀(XRD-6000,SHIMADZU)對LiNiVO4產(chǎn)物及充放電后的LiNiVO4電極進行物相分析。采用X-650 (Hitachi)型掃描電子顯微鏡觀察充放電前后LiNiVO4電極的形貌。
1.3 LiNiVO4電極的制備及其電化學(xué)性能測試
將LiNiVO4粉末、乙炔黑和聚四氟乙烯乳液(PTFE)按8:1:1的質(zhì)量比例均勻混合,搟壓成膜,壓于鋁網(wǎng)集流網(wǎng)上制成正極。以金屬鋰片作負極,Celgard2400作為隔膜,1mol dm–3 LiPF6的EC-DMC溶液(EC與DMC的體積比為1:1)為電解液組裝成CR2016扣式電池,在Land電池測試儀上進行恒流充放電實驗,充放電截止電壓為4.9-3 V,充放電比電流為15 mA
dm–3 LiPF6的EC-DMC溶液(EC與DMC的體積比為1:1)為電解液組裝成CR2016扣式電池,在Land電池測試儀上進行恒流充放電實驗,充放電截止電壓為4.9-3 V,充放電比電流為15 mA g–1。用一小玻璃瓶盛裝電解液并制作成密封性能良好的三電極模擬電池,將此模擬電池在CHI660A電化學(xué)工作站上進行循環(huán)伏安(CV)實驗,掃描范圍為4.9-3 V,掃描速率為50 μV
g–1。用一小玻璃瓶盛裝電解液并制作成密封性能良好的三電極模擬電池,將此模擬電池在CHI660A電化學(xué)工作站上進行循環(huán)伏安(CV)實驗,掃描范圍為4.9-3 V,掃描速率為50 μV s–1。電化學(xué)阻抗譜(EIS)的測試在AUTOLAB PGSTAT12型電化學(xué)工作站(荷蘭ECO CHEMIE)上進行,測試的頻率范圍為100 kHz-10 mHz,電壓激勵振幅為5 mV。為了驗證被LiNiVO4循環(huán)過的LiPF6電解液的功能,采用商品化的LiCoO2為正極與其組裝成CR2016扣式電池進行恒流充放電實驗。所有電池的組裝均在充滿高純氬氣的手套箱(MECAFLEX MECABOX80-1s,瑞士產(chǎn))中進行。
s–1。電化學(xué)阻抗譜(EIS)的測試在AUTOLAB PGSTAT12型電化學(xué)工作站(荷蘭ECO CHEMIE)上進行,測試的頻率范圍為100 kHz-10 mHz,電壓激勵振幅為5 mV。為了驗證被LiNiVO4循環(huán)過的LiPF6電解液的功能,采用商品化的LiCoO2為正極與其組裝成CR2016扣式電池進行恒流充放電實驗。所有電池的組裝均在充滿高純氬氣的手套箱(MECAFLEX MECABOX80-1s,瑞士產(chǎn))中進行。
2 結(jié)果與討論
2.1 LiNiVO4粉末的XRD圖譜分析
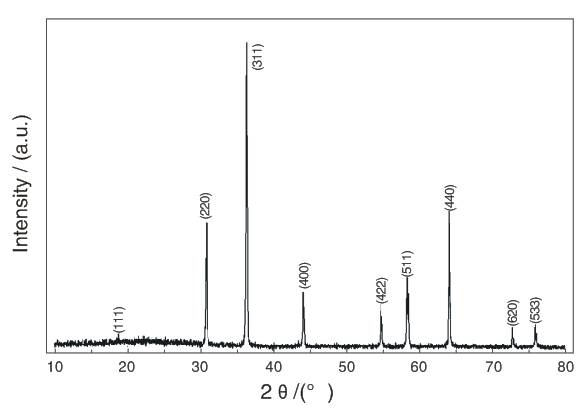
圖 1?? LiNiVO4 粉末的XRD圖譜
圖1 為600 下加熱前驅(qū)體10h得到的LiNiVO4粉末的XRD圖譜。LiNiVO4粉末的XRD圖與LiNiVO4的XRD標準圖譜(JCPDS 73-1636)相一致,屬于立方晶系、Fd-3m空間群,精修XRD得到了的晶胞參數(shù)a=8.2208(1)
下加熱前驅(qū)體10h得到的LiNiVO4粉末的XRD圖譜。LiNiVO4粉末的XRD圖與LiNiVO4的XRD標準圖譜(JCPDS 73-1636)相一致,屬于立方晶系、Fd-3m空間群,精修XRD得到了的晶胞參數(shù)a=8.2208(1) 。與尖晶石LiMn2O4不同的是,LiNiVO4在XRD圖出現(xiàn)了強的(220)衍射峰,而位于2θ約為19.02
。與尖晶石LiMn2O4不同的是,LiNiVO4在XRD圖出現(xiàn)了強的(220)衍射峰,而位于2θ約為19.02 的(111)衍射峰則非常弱。另外,LiNiVO4粉末的(220) 衍射峰與(311)衍射峰的強度比約為0.5,表明流變相反應(yīng)法合成的LiNiVO4粉末具有高的結(jié)晶度[9]。
的(111)衍射峰則非常弱。另外,LiNiVO4粉末的(220) 衍射峰與(311)衍射峰的強度比約為0.5,表明流變相反應(yīng)法合成的LiNiVO4粉末具有高的結(jié)晶度[9]。
2.2 LiNiVO4粉末的電化學(xué)性能表征
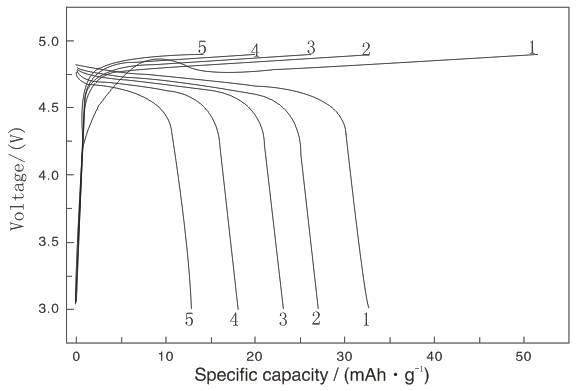
圖2? 為得到的LiNiVO4粉末的充放電曲線
圖2為LiNiVO4粉末在放電速率為15 mA g–1、充放電截止電壓為4.9-3 V時的前五個循環(huán)充放電曲線。由圖可知,LiNiVO4粉末的首次充放電容量為51.5 和32.6 mAh
g–1、充放電截止電壓為4.9-3 V時的前五個循環(huán)充放電曲線。由圖可知,LiNiVO4粉末的首次充放電容量為51.5 和32.6 mAh g–1,首次不可逆容量損失為18.9 mAh
g–1,首次不可逆容量損失為18.9 mAh g–1,但這個放電容量要高于Fey G T K所報道的數(shù)據(jù)[7,8,10]。然而,隨著充放電循環(huán)的進行,LiNiVO4粉末放電容量快速衰減,放電平臺降低,充電平臺升高且充放電平臺縮短,第五個循環(huán)的放電容量已下降至12.9 mAh
g–1,但這個放電容量要高于Fey G T K所報道的數(shù)據(jù)[7,8,10]。然而,隨著充放電循環(huán)的進行,LiNiVO4粉末放電容量快速衰減,放電平臺降低,充電平臺升高且充放電平臺縮短,第五個循環(huán)的放電容量已下降至12.9 mAh g–1。
g–1。

圖3?? LiNiVO4粉末的循環(huán)伏安曲線
圖3為LiNiVO4粉末的前三個CV曲線。從圖上可以看出,LiNiVO4粉末的首次CV曲線分別在4.792和4.665 V處出現(xiàn)了一對氧化還原峰,其歸屬于Ni3+/Ni2+氧化還原電對的單電子轉(zhuǎn)移過程。然而,從第二個循環(huán)起,氧化還原電流峰突然消失,在目前的一些關(guān)于LiNiVO4的CV研究報道中,也出現(xiàn)了第二次循環(huán)時氧化還原峰突然消失或沒有明顯的氧化還原峰的現(xiàn)象[4,11,12],研究者都將其中的原因歸結(jié)為高電壓下電解液發(fā)生了分解,但沒有給出相關(guān)的直接證據(jù)。

圖 4?? 不同循環(huán)次數(shù)下LiNiVO4電極的EIS圖
圖4為 LiNiVO4電極在開路狀態(tài)下和CV測試三個循環(huán)后的EIS圖。LiNiVO4電極在開路狀態(tài)下的EIS圖形由一個高頻區(qū)的半圓和一條低頻區(qū)的直線組成。其中,高頻區(qū)的半圓與電極和電解液表面之間的電荷轉(zhuǎn)移步驟有關(guān),而低頻區(qū)的直線則反映了鋰離子在固態(tài)電極中的擴散情況。經(jīng)過三次循環(huán)后,EIS圖發(fā)生了變化,由高頻和中頻區(qū)的兩個半圓環(huán)以及低頻區(qū)的擴散直線組成。一般來說,第一個半圓代表電極表面SEI膜中的離子傳導(dǎo),第二個半圓代表電子在電極活性物質(zhì)中的擴散,低頻直線表示的是Li+離子在固相中的擴散。顯然,經(jīng)過循環(huán)后LiNiVO4電極的電化學(xué)反應(yīng)阻抗增大,說明進行電化學(xué)反應(yīng)變得比較困難。而當LiNiVO4在高電壓下進行脫嵌鋰時,會發(fā)生溶劑和電解質(zhì)電極表面還原進而生成鈍化膜,反復(fù)循環(huán)可能導(dǎo)致SEI膜厚度增加從而使其阻抗增大。
2.3 LiNiVO4電極的ex-situ XRD圖譜及SEM分析
圖5給出了LiNiVO4電極在充放電循環(huán)三次后的ex-situ XRD圖。循環(huán)后的LiNiVO4電極的XRD譜除了在2θ約為38.69 、44.63
、44.63 、64.95
、64.95 和78.03
和78.03 處出現(xiàn)了四個鋁集流網(wǎng)的特征衍射峰外,其它的衍射峰的位置與圖1所示的XRD譜是一致的,即LiNiVO4在循環(huán)后能夠恢復(fù)為原有的結(jié)構(gòu),同時其各個衍射峰的強度與充放電前相比也沒有發(fā)生大的變化。LiNiVO4電極的ex-situ XRD結(jié)果說明LiNiVO4在循環(huán)過程中的結(jié)構(gòu)是可逆的。
處出現(xiàn)了四個鋁集流網(wǎng)的特征衍射峰外,其它的衍射峰的位置與圖1所示的XRD譜是一致的,即LiNiVO4在循環(huán)后能夠恢復(fù)為原有的結(jié)構(gòu),同時其各個衍射峰的強度與充放電前相比也沒有發(fā)生大的變化。LiNiVO4電極的ex-situ XRD結(jié)果說明LiNiVO4在循環(huán)過程中的結(jié)構(gòu)是可逆的。
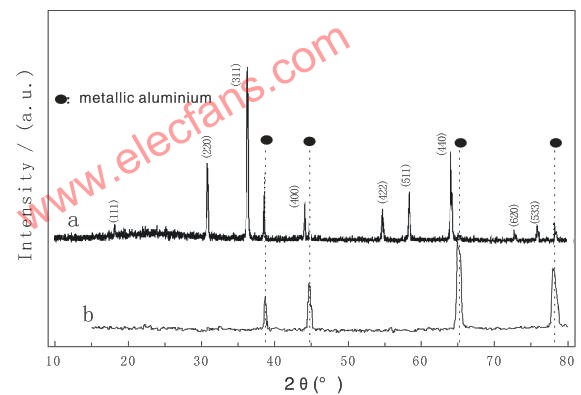
圖5?? 循環(huán)三次后的LiNiVO4電極的非現(xiàn)場XRD圖 (a) LiNiVO4電極;(b) 鋁集流網(wǎng)

圖6 LiNiVO4電極膜的SEM照片 (a)原始的;(b)循環(huán)三次的
圖6進一步給出了原始的LiNiVO4電極膜和經(jīng)過CV循環(huán)三次后的電極膜的SEM照片。與原始的LiNiVO4電極膜相比,循環(huán)三次后的LiNiVO4電極表面出現(xiàn)了許多“裂縫”,有的寬度達到1.5 μm左右,并且在“裂縫”處附著了許多纖維狀的細絲,這極有可能是“裂縫”形成的同時撕裂了LiNiVO4電極中粘結(jié)劑PTFE的網(wǎng)絡(luò)結(jié)構(gòu)。LiNiVO4電極表面的開裂現(xiàn)象所造成的一個直接后果就是“裂縫”處的活性顆粒難以充分接觸,從而使局部的活性顆粒無法進行電化學(xué)反應(yīng)。而且,LiNiVO4電極的表面開裂極有可能導(dǎo)致其表面SEI膜的破壞。這樣,在每一次氧化還原過程中,LiNiVO4電極與LiPF6電解液之間都會存在局部的反應(yīng),以修補被破壞的SEI膜。這種反應(yīng)不僅會消耗LiPF6電解液,而且會使得這層非電化學(xué)活性的表面膜逐漸增厚,增加了電化學(xué)反應(yīng)的電阻[13],從而使電極的放電容量急劇下降。
2.4 LiPF6電解液的功能驗證

圖7 LiCoO2在被LiNiVO4循環(huán)不同次數(shù)的LiPF6電解液中的首次充放電曲線
許多研究者都將LiNiVO4較差的電化學(xué)性能歸因于在高電壓下電解液發(fā)生了分解,為了驗證這一情況,我們將經(jīng)過CV測試的三電極模擬電池放入手套箱中,取出其中的LiPF6電解液和新鮮的LiPF6電解液分別與商品化的LiCoO2正極材料組裝扣式電池進充放電測試。圖7給出了商品化的LiCoO2于30 mA g–1下、4.4-3 V范圍內(nèi)在不同的LiPF6電解液中的首次充放電曲線。從圖上可以看出,LiCoO2在新鮮的LiPF6電解液中首次充放電容量分別為181.5和166.1 mAh
g–1下、4.4-3 V范圍內(nèi)在不同的LiPF6電解液中的首次充放電曲線。從圖上可以看出,LiCoO2在新鮮的LiPF6電解液中首次充放電容量分別為181.5和166.1 mAh g–1;在經(jīng)歷了一次CV實驗的LiPF6電解液中,LiCoO2的首次充放電容量僅為129.8和98.1 mAh
g–1;在經(jīng)歷了一次CV實驗的LiPF6電解液中,LiCoO2的首次充放電容量僅為129.8和98.1 mAh g–1,并且充電平臺升高,這極有可能暗示著電解液此時已經(jīng)發(fā)生了分解;為了進一步說明這一問題,我們采用經(jīng)歷了三次CV掃描的LiPF6電解液進行同樣的實驗,發(fā)現(xiàn)LiCoO2的充放電曲線已變得畸形,放電容量僅為26.5 mAh
g–1,并且充電平臺升高,這極有可能暗示著電解液此時已經(jīng)發(fā)生了分解;為了進一步說明這一問題,我們采用經(jīng)歷了三次CV掃描的LiPF6電解液進行同樣的實驗,發(fā)現(xiàn)LiCoO2的充放電曲線已變得畸形,放電容量僅為26.5 mAh g–1,這充分說明此時電解液已經(jīng)基本喪失了離子傳導(dǎo)功能。
g–1,這充分說明此時電解液已經(jīng)基本喪失了離子傳導(dǎo)功能。
由此可以預(yù)見,LiNiVO4作為鋰離子電池正極材料的容量衰減并非是由于其結(jié)構(gòu)遭到了破壞(圖5已證實圖LiNiVO4結(jié)構(gòu)的可逆性),而是源自LiNiVO4與電解液的相互作用對雙方的負面影響。一方面,LiNiVO4在高電壓下能夠催化分解電解質(zhì)溶液。Fey G T K曾采用抗氧化性較強的LiBF4/DMC+PC+EC(體積比為:66:17:17)作為電解液,LiNiVO4的首次放電容量可達到45 mAh g–1 [1]。而在隨后的研究中,由于使用LiPF6電解液,盡管采用了一些軟化學(xué)方法合成LiNiVO4,但LiNiVO4的放電容量沒能超過30 mAhg–1 [7,8,10]。另一方面,電解質(zhì)溶液的分解產(chǎn)物(如聚碳酸酯、烷基碳酸鹽、氟化鋰等)及LiNiVO4電極表面的開裂現(xiàn)象會增大SEI膜和電化學(xué)反應(yīng)的阻抗,使LiNiVO4喪失電化學(xué)活性,從而導(dǎo)致其容量衰減。
g–1 [1]。而在隨后的研究中,由于使用LiPF6電解液,盡管采用了一些軟化學(xué)方法合成LiNiVO4,但LiNiVO4的放電容量沒能超過30 mAhg–1 [7,8,10]。另一方面,電解質(zhì)溶液的分解產(chǎn)物(如聚碳酸酯、烷基碳酸鹽、氟化鋰等)及LiNiVO4電極表面的開裂現(xiàn)象會增大SEI膜和電化學(xué)反應(yīng)的阻抗,使LiNiVO4喪失電化學(xué)活性,從而導(dǎo)致其容量衰減。
3 結(jié)論
通過流變相反應(yīng)法成功制備了純相的高電壓鋰離子電池正極材料LiNiVO4。Ex-situ XRD的研究結(jié)果表明了LiNiVO4在鋰離子嵌入和脫出過程中的結(jié)構(gòu)是可逆的;而電解質(zhì)溶液在高電壓下的分解和LiNiVO4/電解質(zhì)溶液界面差相容性是其容量衰減的主要原因。因此,尋找電化學(xué)窗口寬、抗氧化能力強的電解質(zhì)溶液以及改善LiNiVO4/電解質(zhì)溶液界面相容性是提高高電壓鋰離子電池正極材料LiNiVO4電化學(xué)性能的關(guān)鍵。
參考文獻
[1] 李宇展, 任慢慢, 吳青端等.鋰離子蓄電池釩系正極材料的研究進展[J].電源技術(shù),2005, 29: 24-127