錳基化合物在化學電源中的優勢與潛力
摘要:當前化學電源中的正極材料大都使用過渡元素氧化物或其衍生物,而錳基材料更具優勢。在化學電源中應用最廣的是MnO2,如鋅/碳電池,堿錳電池,可充堿錳電池。對各類晶型MnO2結構研究較為透澈,又可互相轉變,并可制備Li-Mn-O化合物:如尖晶石型LiMn2O4和層狀結構的LiMnO2和層狀-層狀復合物是動力鋰離子電池的候選材料。與LiCoO2相比較,Mn-基材料的優勢是:資源豐富,價格低廉,環境友好,安全性強。
關鍵詞:MnO2電池;層狀鋰-錳-氧化物;尖晶石型鋰-錳-氧化物
當前實用化學電源中的陰極,幾乎都使用金屬氧化物或其衍生物作為活性材料。而眾多的電池體系中,以二氧化錳作為活性材料的最多。如一次電池中的鋅碳電池、堿錳電池、鋰-錳電池。貯備電池水激活鋅錳電池中的MnO2。二次電池中有可充堿錳電池,鋰合金可充電池的MnO2,以及在鋰離子電池正在大力研發的錳基衍生物如LiMnO2、Li2MnO3、LiMn2O4等。
1 MnO2作為電極材料的優勢
MnO2是一種多晶型材料,隨著制備條件的不同,可有不同的晶型、形態、粒徑、孔隙率、比表面積,而適用于不同的應用場合。既可用于一次電池中,又可用于二次電池中。既可用于水溶液(弱酸性或堿性)中,也可用于有機溶劑中,既可作為可充電池的正極活性材料,又可作為超級電容器或電化學電容器的活性材料。其應用之廣,在諸多電池正極活性材料中是少見的,而且MnO2的研究跟著應用的需求,性能也在與時具進。
1.1 MnO2的晶型彼此間可互相轉化
二氧化錳的晶體結構可分為三大類,存在5種主晶和30余種次晶。但其基本結構單元是由1個錳原子與6個氧原子配位組成的六方密堆積或立方密堆積結構[MnO6]八面體,八面體與鄰近的八面體共棱或共角頂即形成變化多端的復雜網絡,這些網絡又可容納各種不同的陽離子或水分子,就造成多種多樣不同類型的晶體結構。在結構中把未被占有的八面體晶格點形成平行于C軸的空隙(或空位)定義為隧道。隧道用T[m×n]表示其大小(T為隧道結構,m是隧道高度,n為隧道寬度)。據此可將MnO2的晶體結構分為三大類:即一維隧道結構[1×1]的β- MnO2(軟錳礦);由軟錳礦和斜方錳礦[1×2]交互生長構成的γ- MnO2,和[2×2]的α- MnO2(堿硬錳礦、隱鉀錳礦、鉛硬錳礦、水鈉錳礦、雜硬錳礦、欽鎂錳礦等具有不同陽離子);具有層狀結構的二維隧道結構的δ-MnO2。層間含有不同陽離子或水分子而有層間距不同的次晶(如合成鈉水錳礦和水錳礦),以及衍生物Li-Mn-O層狀物(LiMnO2、Li2MnO3…);具有網狀結構的三維隧道的λ-MnO2及Li-Mn-O的衍生物的LiMn2O4等等。
而這些不同晶體結構的MnO2及其衍生物在一定條件下發生晶型互變如圖1所示。圖中晶型的互變轉化條件參見已發表的文章[1]。
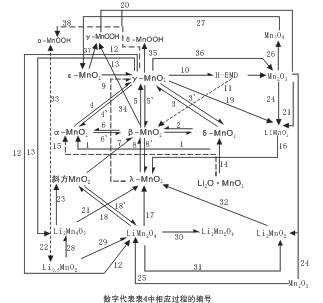
圖1? 二氧化錳及其相關鋰錳氧化物晶體結構互變關系
由圖1可以看出由α、β、γ、δ-MnO2與斜方MnO2,在一定條件下可轉變為尖晶石結構λ-MnO2、LiMn2O4、Li2Mn4O9等,由γ-MnO2可轉化為LiMnO2,因此又聯系到鋰錳氧化物間的互變,而后者正是鋰離子電池可供選擇的正極活性材料,從而大大拓寬了錳基材料應用的空間,而且這些錳基氧化物活性材料還比其金屬氧化物材料具有獨特的優勢。
1.2 資源豐富
錳在地殼中貯量居第12位,在過渡金屬元素中只有Fe的貯量超過錳。資源豐富在考慮用于混合電動車(HEV)和電動車(EV)的電源正極材料是一個重要因素。錳礦生產為MnO2有成熟的技術(包括電解法和化學法),這也是MnO2正極活性材料具有的優勢。
1.3 價格低廉
在一次電池中或二次電池中,很少有比MnO2價格便宜的正極活性材料。從原材料價格,2008年進口的天然錳礦(含錳量43%~45%)到岸價約為590美元/噸(約合4000元/噸),同期生產的電解二氧化錳(EMD)價格約10500元/噸,而進口鈷價最高約90萬元/噸,因此合成MnO2只有合成LiCoO2的價格的1%左右。而在鋰離子電池中的Li-Mn-O材料比起Li-Co-O也要便宜得多。
1.4安全性能優良
用于電池特別是在HEV或EV中在過充或過放時,錳基正極材料(特別是MnO2)要比LiCoO2或LiNiO2等材料發熱量要少,因而從過充到Mn4+時有較其他到Co4+安全或Ni4+的穩定性要好。
1.5 環境友好
錳基材料比Co基材料的毒性來說,是無毒材料,在對環境友好來說也有重要的意義。
2 近十年MnO2電池取得巨大進步
2.1 完成了鋅錳電池從鋅碳電池、紙板電池到堿錳電池的無汞化的過渡。我國是一次電池的生產大國,產量占世界50%以上。2007年產量達300.7億只(堿錳電池82億只);2008年1月-7月產量達225億只;10年前我國堿錳電池產量僅有5億只,2008年前7個月產量近90億只,在這個發展過程中并實現了無汞化。這得益于我國電解二氧化錳(EMD)的技術的進步與產量的激增。紙板電池得益于普通EMD的加入,而無汞化堿錳電池得益于專用級EMD的增長。
2007年鋅碳電池級EMD產量達78337噸,而堿錳電池級EMD產量達81542噸,占世界EMD產量的41%以上。
隨著科技的進步,高技術消費電子設備大量進入市場,大多是需要高功率的堿錳電池,這又推動了高功率堿錳電池的研發。
2.2 高功率堿錳電池的成就[1-5]
(1)要有高功率堿錳電池,首先要具有高功率放電能力的晶體結構為γ- MnO2型EMD[6]。而且這種EMD需要從粒徑、粒度分布、比表面、微孔面積、微孔體積、孔隙率、孔徑、最高開路電壓等各種因素的協調,就要有適宜的電解制備條件:改變與調整原來常法的電解條件,使硫酸、硫酸錳的濃度及其濃度比最佳化,電流密度的控制,溫度的選擇范圍,以及三者的協商[7, 8]。此外還有水熱法,可以控制電解條件,可分別制得不同的α、β、γ、δ-MnO2,分別用于堿錳電池或非水溶液中的鋰錳電池[9];以及利用水熱法電解,可生產出有適宜的孔結構的高功率EMD[10]。
(2)評價高功率EMD的方法:
Bowden等[11, 12]利用階躍電位電化學譜(SPECS)測得EMD樣品的功率電位圖,并將圖上高電位(1.45V)處的峰值功率與低電位(1.1V)處的峰值功率之比定義為功率系數。功率系數越大,表示高電位放電時間越長,功率系數就越大,EMD的高功率性能的γ-MnO2功率系數最好在4.0以上,實際上,定義功率系數是評價EMD的高電壓段放電性能。
利用電子順磁共振測定γ-MnO2參數法。因為γ-MnO2并非化學計量化合物,具有缺陷結構,恰當的化學式為: 。在電子順磁共振(EPR)譜中,有兩種信號A和信號B。若MnO2中Mn均為4價,則B的信號寬度為△B0=430mt;由于存在Mn3+,偶極-偶極的交換收縮改變成“Zener”雙交換,則信號寬度改變。△B0越大,Mn3+/Mn4+越大,即OH基越多,缺陷結構存在的無序性越大,嵌入H+的容量也越大[13]。
。在電子順磁共振(EPR)譜中,有兩種信號A和信號B。若MnO2中Mn均為4價,則B的信號寬度為△B0=430mt;由于存在Mn3+,偶極-偶極的交換收縮改變成“Zener”雙交換,則信號寬度改變。△B0越大,Mn3+/Mn4+越大,即OH基越多,缺陷結構存在的無序性越大,嵌入H+的容量也越大[13]。
當然高功率堿錳電池是一種綜合工藝,還需要有高功率的無汞鋅粉[14],電解液的組成和濃度以及在電池中的含量;各種添加劑的應用,導電材料的選擇;降低內阻特別是各部件間的接觸電阻(尤其是在EMD、石墨和電液三相界面的有效接觸,因此涉及正極成形的工藝如混料造粒,打環以及MnO2和石墨的粒徑的配合,形成正極結構孔隙率,以提供質子-電子對定位化有暢通的通道[15]);根據不同型號的電池有相應的優化配方等等。進一步提高其性能,仍有較大的研發空間。
近來報導,用低溫水熱法制得的直徑為1~4μm呈海膽狀的α-MnO2用于堿錳電池中放電率為50、200、500mA/g放電至0.6V,放電容量分別達280.5、185.9和168.2mAh/g;而對比的EMD的放電容量分別為254.9、121.1和90.2mAh/g,說明在高電流密度下α-MnO2的放電性能較好[16]。λ-MnO2在堿錳電池中的應用也取得了進展[17]。
2.3 可充堿錳(RAM)電池
從節約一次資源,減少廢棄電池對環境造成的局部污染兩方面考慮,研發RAM電池是人們長期關注的課題。早在20世紀60年代末就出現了市售RAM電池,但由于二氧化錳電極的可逆性較差,隨著充放循環工作電壓迅速下降,而且因爬堿嚴重而作罷。經過近30年的努力,至20世紀90年代末,國內外又興起RAM電池生產熱潮,國外產量達數億只。主要是通過對EMD(γ-MnO2)的摻雜改性[18, 19],加入金屬氧化物如Bi2O3,TiO2(銳鈦型),PbO、PbO2、SnO2等(加入的方法包括物理法、化學法和電化學法改性),以及基本弄清了γ-MnO2可逆性差不宜進行2e的反應充放電機理[20, 21],在le反應γ-MnO2放電生成的是可溶MnOOH,而到了2e/2H+階段充放電生成了非活性的Mn3O4 ,因而若充放電限制在le/1H+內(最好在0.5e/0.5H+),加入有效的改性摻雜物,也可避免2e/2H+階段Mn3O4的生成[22]。
當然,生產整個RAM電池,還需要負極活性材料,隔膜、電液濃度,特別是充放電的方法的選定。
關于添加劑,近來有了新的進展。我們過去曾用磁場處理γ-MnO2,發現在高電壓區的放電量有大的改進[23]。近來通過加入永磁材料粒子如SmCo5、SmCo17等一系列鐵磁、亞鐵磁、超導材料及其組合物來改性EMD[24]。發現磁性粒子在正極內存在,影響了EMD還原態的晶體結構,同樣可避免充放電過程Mn3O4的生成,從而增大了MnO2的放電容量,例如在le/1H+均相還原中,是未加磁性粒子的MnO2放電容量1.5倍;而到2e/2H+區,總放電量是后者的2.0倍。而用于RAM電池中,磁性粒子SmCo17電極15次循環累積充電容量為3.96Ah/g(le/1H+區),放電容量累積為4.67Ah/g,初始放電容量達0.546Ah/g;而未處理的分別為2.144Ah/g,1.865Ah/g和0.466Ah/g;因而加有磁性粒子對電流效率提高和放電容量,循環性都極為有益。
與其他二次電池的活性材料一樣,除了摻雜改性外,還可進行表面修飾和包覆,如使用膠體超細石墨包覆MnO2,用于AA型RAM電池中,若包覆量占正極的3.2wt%,內阻為88.2Ω,包覆量0.6wt%內阻為174.2Ω;而未包覆的內阻卻是535.2Ω[25]。由于超細碳粒徑只有150nm,包覆在活性材料表面,可減少正極中導電石墨的含量,增大了MnO2的裝填量,也增大了電池的放電容量[26]。
近年來,在改善RAM電池的可充性,還提出了用脈沖電流電解Mn2+代替直流電解的方法,并降低槽溫(60℃-80℃),以獲取不同的缺陷結構(在MnOn中的n不同)有不同的含水量,發現n在1.957~1.976時循環性最佳[27]。
應該說RAM電池還有發展的空間,比起一次堿錳電池,性價比明顯優越得多。
2.4 各種晶型MnO2在堿液中的可充性
各種晶型的MnO2在堿液中的可充性是不同的。即使同為EMD或CMD同是γ-型MnO2,不同樣品其可充性不相同[28],因為γ-MnO2的缺陷結構不一定相同。γ-MnO2的缺陷結構有兩類,一類是Dewolf缺陷,相應于金紅石型的β-MnO2在斜方錳礦中交互生長的濃度分數(Pt表示);另一類是孿晶缺陷,是共棱八面體鏈鋸齒形生長比率(Mt或Tw表示)。在水溶液中嵌H+,最佳的是缺陷結構在46%≤Pt≤50%,即交互生長的無序性最大越好。同是γ-MnO2,其缺陷Pt與Mt不同,其嵌入H+的動力學有差異而不同,晶型結構不同,或由于隧道大小不同或由于含水量和雜離子的不同也會影響H+/e對的傳輸,其反應機理也有異,如δ-MnO2充放電過程的均相只在δ-MnO2→MnOOH0.60,嵌入H+超過0.6,即在充放電過程生成Mn3O4或Mn2O3;且由于層間的收縮與層內的膨脹不同,而影響其結構的穩定性。λ-MnO2是[1×1]隧道形成的三維網絡結構,H+的嵌脫不易,因而可充性差。
2.5 MnO2在非水溶液中的可充性
γ-MnO2在非水溶液嵌脫Li+與在水溶液中嵌脫H+恰恰相反,一般應有最大的有序,即其缺陷結構參數小,即Mt→0,或Pt→0。例如在Li/MnO2電池中,將EMD加熱處理脫水成為HEMD,生成β/γ,接近β-MnO2而呈有序化[29]。
α-MnO2在嵌Li電池中是研究的熱點之一,因為α-MnO2是[2×2]隧道結構,甚或[3×3]隧道結構,有嵌Li+的可能。為了嵌Li+有一系列的制備方法:早期的Mn2+的氧化,Mn2O3在H2SO4中的岐化;Li2MnO3在酸中處理,目的是制得不含K+、Ba2+等雜離子而含水量大的α-MnO2 0.15H2O,因為它具有高的熱穩定性(490℃)。顯然水分子是取代雜離子在隧道中穩定α-MnO2的結構。而NH3分子與水分子大小相近,鍵合性質相似,因而用NH3氣在低溫下處理而形成在隧道中由NH3與H+形成NH4+,穩定α-MnO2結構的NH3 α-MnO2。進而用水化型α-MnO2 0.15H2O與LiOH混合、干燥、燒結(300℃)形成Li2O-α- MnO2。再用NH3與Li2O同時混合形成Li2O-NH3-α-MnO2。這些不同的隧道分子的α-MnO2,在3.8-2.0V間,電解液為1M LiPF6/EC:DMC(1:1)的Li/MnO2電池中,π=0.2mA/cm2時,放電初始容量均超過了220mAh/g,且在循環20次時,Li2O-NH3-α-MnO2,Li2O-α-MnO2,NH3α-MnO2與α-MnO2 0.15H2O的放電容量分別為223、188、167和123 mAh/g。前面三種α-MnO2的放電曲線并無下降趨勢,即大大改善了α-MnO2的充放循環性能[30, 31]。
層狀結構δ-MnO2可由Li2MnO3析出而得到Li2O,組成Li/δ-MnO2電池,在i=0.2mA/cm2,3.6-2.4V間充放電,其初始容量約200 mAh/g,而在第8次循環放電容量只有140 mAh/g左右,說明容量衰減迅速[32]。
對三維網狀結構的λ-MnO2,美國曾擬做Li/λ-MnO2電池取代Li/SO2電池在部隊中使用,因為它有較好的低溫性能,而且在高電位區優于Li/HEMD(β/γ-MnO2)電池,特別是4V平臺對低溫有利[33]。但作為二次Li電池λ-MnO2放電,易發生多相變化而未見實用化。
2.6 MnO2與錳氧化物的應用拓展
由圖1可以看出,各類MnO2的互變以及各種錳基原料如Mn2+化合物、MnO2、Mn2O3、Mn3O4,都可制備EV和HEV的鋰離子電池正極活性材料:如尖石晶型,層狀,以及層狀化合物的復合材料,引人關注。
主要錳基鋰離子電池正極材料基本的物理參數和電化學性能如表2所示。錳基化合物的制備方法如表3所示
表2 錳基鋰離子電池正極活性材料的物理參數和電化學性能
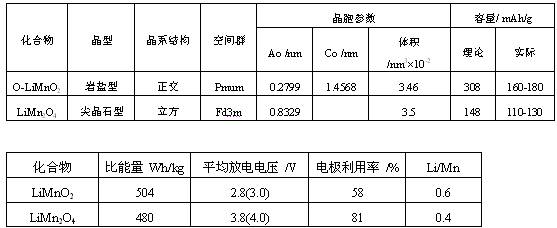
表3 Mn基化合物的制備方法

材料必需具備單一的物相,良好的一致性,均勻的顆粒形態,并有亞微細粒甚或超微的粒子分布,以及大的比表面積。影響其本征性質的是粒子的形狀,尺寸(粒徑),取向、空隙、晶粒間界有大的原子分數。這就需要調整制備的條件。同樣的制備方法可能有不同的效果。
2.6.1 LiMn2O4的優勢與存在問題
LiMn2O4具有尖晶石型三維網絡結構。Li+占據四面體(8a)的晶格點,Mn離子占據八面體(16d)的晶格點,氧處于32e晶格點呈敞開式立方密堆積排列。在Mn2O4構架中晶隙空間可視為四面體8a位置起著Li+遷移脫嵌的通道。因而適宜用作鋰離子正極活性材料的本征性質,而且原料豐富,成本低廉,對環境友好,尤其是熱穩定性好,安全性佳,是具有發展潛力的鋰離子正極材料。但也由于Mn基材料的固有性質,在充放電循環過程中的容量衰減主要由于Mn的存在而產生。如Mn2+在非水電解液中的溶解損失;在高電位下,由于Mn3+的岐化Jahn-Teller效應引起結構不穩定的損失;在高溫下,PF與水發生反應形成的HF對電極的侵蝕;隨著充放循環Mn由16d位置移向8a位置堵塞了Li+通道造成的損失以及產生結構無序化的損失。這些都與充放過程結構發生變化有關。解決容量衰減的方法大致有:改善合成材料的方法(如表2所示的各種方法)和條件(特別是燒結溫度與時間);對體相的摻雜或取代部分Mn;對材料表面的改性,修飾和處理;或者既取代又表面改性。
(1)體相摻雜取代
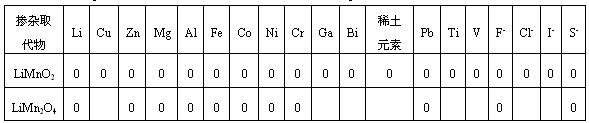
摻雜取代的方式有單元或多元陽離子的取代;單元或多元陰離子的取代;陽離子取代Mn和陰離子取代O同時進行。取代摻雜是一種折衷的選擇,有利也會有弊。有的能改善循環,卻可能要降低初始容量;有的陽離子取代可提高容量,卻可使循環性能變差。多元取代陰陽離子取代可能帶來合成的困難或條件的苛刻。所有的摻雜取代都有最適量,否則會帶來結構的變化。對Mn-基化合物體相改性取代摻雜物如表4所示。
表4 摻雜Mn-基正極活性材料的摻雜取代元素
對摻雜改性元素的選擇大致有以下的規律:(1)摻入元素(M)(如Co、Ni、Cr)與O的結合能M-O要比Mn-O的結合能大,使晶胞收縮,縮短M-O鍵長,提高了LiMn2O4的尖晶石結構的穩定性。但還有不同的特性,如摻Co還能提高Li在體相的擴散系數,有助循環性能的提高;如摻Ni優選出取代量為0.5即構成LiNi0.5Mn1.5O4不同制備方法都可使初始容量達135-140 mAh/g,循環數在40-60次范圍內,容量損失在0~4.5%之間;當取代量x在0.05≤x<0.5之間也可達130 mAh/g左右。循環在100次以上容量衰減也只在2~5%之間,這是因為Ni-O鍵能大,弱化了Li-O鍵的結合能,降低了Li+擴散的內阻。(2)摻雜元素M-O的鍵能也與摻雜取代元素的離子半徑有關。rMn3+=0.0645nm,而rCr3+=0.0615nm,rTi4+=0.0605nm,Cr與Ti的離子半徑均小于Mn3+的離子半徑,因而會使晶胞收縮,結構穩定在循環中不易形變,提高了循環性能,但一般初始容量會降低一些。因此一般要提高循環性能,就要選擇與Mn離子半徑相近或稍小的金屬離子。(3)要抑制材料的Jahn-Teller效應,一般要選擇+2或+3價的離子,主要是提高LiMn2O4中Mn的平均價態。但若既要提高循環性能,又有好的比容量,主要應制備單一的物相[34]。Al3+,Ga3+,Fe3+的離子半徑與Mn3+相同或相近,但均既占據四面體8a晶格點位置又占據八面體16d晶格點位置,而形成反尖晶石離子LiMn5O8存在,但只要M摻雜量最佳化,也有利循環性的提高[35]。(4)陰離子(F-)取代一般負電性比氧大,吸電子能力強,降低Mn在電液中溶解,使LiMn2O4的初始容量提高,但降低了Mn的價態,容易發生Jahn-Teller效應,循環性能會變壞,因此一般用F取代氧,必須有適應的陽離子取代Mn加以協調。
上述原則是以單元取代而言,它們的不足可以用多元取代或摻雜來加以改善,例如Ni取代成效顯著,但未見高倍率的報導。近來用不同的合成法,用Mg取代部分Ni構成LiMgδNi0.5-δMn1.5O4(δ=0.07)其理論容量為148.7mAh/g,非常接近未加任何取代的理論容量148.2mAh/g。結果表明Li/LiMg0.07Ni0.43Mn1.5O4在3.5-4.8V間0.2C放電,用固相法,溶膠凝膠法和干凝膠法制得的樣品的初始容量分別為116、108和106 mAh/g,1C下分別為104.4、97和95.4 mAh/g;30次循環后以5C率放電分別為70、64.8和74.2 mAh/g,表明倍率能力良好[36]。又如Ti取代Mn,以電解法先制得Ti-EMD,再用固相反應法制得Li0.973Ti0.045Mn1.893O4在2.5-4.6V間放電初始容量達206mAh/g,循環40次后容量保持率為20%,貯存3個月初始容量可達144mAh/g,40次循環后容量保持率為14%;而貯存電池在4.3-3.4V內的充放循環約150次后其容量保持率達94.7%。即二元取代既有好的初始容量,循環性,更有良好貯存性能[37]。
不少摻雜取代的離子(無論是+2價或高價離子)在室溫下有不錯的循環性,但是在高溫(55℃)高電位(5V)下容量衰減快,循環性下降:一是引起材料的氧的缺陷,一是引進入16d位置使混亂度增大。但摻Cu構成LiCuxMn2-XO4(0.025≤x≤0.1)范圍內如x=0.1時,在3-5V電壓下初始容量達130mAh/g,隨后的循環可穩定在120mAh/g[38]。5V級的材料早已做過研究如LiCu0.5Mn1.5O4和LiNixCu0.5-xMn1.5O2,它們的優點是使用高電位負極時整個電池也有高工作電壓,但仍受到電解液的限制[39]。
對LiMn2O4的體相改性,雖然作了大量的研究,取得了一些進展,但對容量衰減的機理并未形成統一的認識,有些實驗得出不同的結論,尤其是高功率大電流充放電還未有很好的解決。
(2)LiMn2O4的表面改性。
對LiMn2O4的表面改性應根據應用的要求,或為了提高材料表面電子電導率(如碳包覆),或為了防止材料表面受到電液的侵蝕溶解(如Al2O3包覆),或為了提高表面的孔隙率以適應高倍率充放電以延長循環性能,或為了除去表面膜層提高活性材料的利用率[40]。
選擇的包覆物應在表面形成均勻的包覆薄層,包覆過程的溫度不能破壞材料的結構,包覆方法要盡可能的簡便。已經研究過的Mn-基材料的包覆物有單質,金屬氧化物或其混合物,無機鹽,有機物如表5所示。
表5 Mn-基正極活性材料的包覆物
如表5所示,用單質C、Ag等包覆LiMn2O4主要是提高材料的表面電子電導率,鍍Ni可提高電極的高溫性能[41],用金屬氧化物是在材料表面引入M-O(M為金屬元素)鍵,抑制氧的活性和Mn的溶解,提高材料的循環性能。ZrO2的包覆還可提高放電容量[42]。鹽類(如Li2CO3等)的包覆可抑制電液中產生的HF對電極材料的侵蝕。LiBO2(LBO)包覆,具有較好的離子電導率,減小比表面積,減緩HF的侵蝕,且在高電壓下有效抑制電極與電液的接觸,降低電液的分解[43]。LiCoO2包覆一方面降低了與LiMn2O4電液的接觸,抑制了材料的溶解,而且Co3+可進入16d晶格點位,不但抑制了Jahn-Tellen效應,也減少了Mn3+的岐化,既提高了電極的比容量,也改善了電池的可逆容量[44]。有機物如乙酰丙酮處理LiMn2O4可與其表面上Mn空軌道成鍵,使之不再對電液分解起催化作用,抑制了電液的分解,在發生配位成鍵的同時,還可溶解一些Mn離子形成非電活性的Li2MnO3,因而也可防HF的侵蝕。
LiMn2O4本身以及與電液的熱穩定性不一樣。隨著電池荷電態的不同引起開始反應溫度與反應熱不同。在4.3V充電時,反應熱達-408.2KJ/mol。此時的反應溫度為151℃[45]。雖然遠低于本身在4.3V的240℃,也是較為安全的。
據報導Davidson等合成了LiMn1.5Ni0.5O4,以2.2C放電容量超過了100mAh/g,循環100次后,容量沒有明顯的衰減,還可進行11C放電[46]。
此外,美國阿貢國家實驗室研發了以Li4Ti5O12為負極,LiMn2O4為正極的鋰離子電池,具有極高的功率密度,壽命和安全性。循環3000次后容量保持率達80%;50C放電容量保持率在低電流放電容量的90%[47],而且證明了尖晶型的Li4Ti5O12 LiMn2O4鋰離子電池是最安全的體系[48]。這些指標已接近或達到混合電動車的要求。
2.6.2 層狀LiMnO2
LiMnO2有多種晶型。屬層狀結構的有α-NaFeO2型的單斜m-LiMnO2和巖鹽型呈正方的O-LiMnO2。在熱力學平衡條件下,O-LiMnO2比m-LiMnO2穩定些,前者的理論容量達308mAh/g,后者則為285mAh/g,都比LiCoO2要高。但由于Mn3+ 3d電子能帶分布(t2geg)的反鐵磁相互作用,引起Jahn-Teller效應。使氧的排列發生畸變,難以形成理想的密堆積,影響結構的穩定。在充放電的過程,物相轉變為尖晶石型結構。由于層狀LiMnO2和尖晶石型LiMn2O4的Mn基化合物結構的變化,引起容量的衰減,都是Mn為核心造成,不同的是前者Mn為3價,而后者的Mn平均氧化態為3.5價。因此,兩者的制備方法,對體相的改性和表面的處理修飾,均是大同而有異。若部分Mn被Ni或Co取代,結構呈O2型(O表示八面體氧配位,2表示兩層,每單元晶胞為MO2),即結構變穩些,可抑制相變。隨著制備方法和取代離子的不同,還有O3型結構即為氧八面體配位形成3層,或形成氧四面體配位的T2型,或菱形Na+配位的P2型結構。P2型轉化為O2型只需MO間互相滑移即可,即是離子交換法的理論根據。在電化學充放電過程,T2也可轉變為O2型。如取代形成富Li并Ni取代的層狀物,會構成四面體氧配位的T2型結構,這也是在層狀LiMnO2中Ni取代Mn成為研究熱點,并具有良好的容量和循環性的原因。
關于LiMnO2的制備方法,體相改性,和表面修飾已分別在表3、4和5中列出。在體相改性中,值得一提的是構成富鋰(或過嵌鋰)和缺鋰的兩類化合物。鋰的嵌入,總的來說取代部分Mn,占據Li層中四面體位置,有利于保持結構的穩定性。當過嵌鋰,可提高Mn的平均氧化態。并抑制Jahn-Teller效應。早期Lu等[49]合成了Li[NixLi(1/3-2x/3)Mn(2/3-x/3)]O2,x=5/12時在3.0-4.4V間以30 mAh/g電流密度放電,容量達150 mAh/g,且安全性優于LiCoO2,容量和循環性都得到提高[50]。Li(Li0.2Ni0.1Co0.2Mn0.5)O2可視為兩種層狀物的固溶體,也可視為過嵌或取代的層狀化合物或超晶格結構的化合物,初始容量可達234mAh/g,40次循環后容量保持率達98.3%。過嵌鋰要適中,或有利于容量,或有利于循環性能的改進。有的富鋰如Li[Lix(Ni0.5Mn0.5]O2發現混排度低于LiNi0.5Mn0.5O2;而Li[LixNi1/3Co1/3Mn1/3]O2(0.03≤x≤0.17)比Li(Ni1/3Co1/3Mn1/3)O2的混排度要小,有序性增大,對容量有利。
對缺Li的可轉化為O2型,O2-Li2/3+x(Co0.15Mn0.85)O2(x=0, 1/3)兩種化合物當x=1/3即為化學計量化合物,當x=0即為缺Li的層狀化合物可轉化為O2型。O2-Li2/3+x(Co0.15Mn0.85)O2在電流密度5 mA/g時可逆容量均為180 mAh/g,在電流密度140 mA/g即1C下,兩種均為100 mAh/g(在2.5-4.6V間)。由EIS研究表明在循環10~15次后O2(Li+x)的容量衰減增大,這與表面膜形成與體相的電阻增大一致。隨后的循環(≤6)次,鋰離子擴散系數:DLi為2×10-11~10×10-11cm2/S,而O2(Li)為0.5×10-10~3×10-10 cm2/S;即化合物的容量衰減,除了界面動力學外,也與DLi值(即體相內)的變化有關[51]。說明缺鋰化合物對抵抗容量衰減有利。此外由Na0.7(Ni1/6Mn5/6)O2通過離子交換法制得Li0.7(Ni1/6Mn5/6)O2是缺鋰化合物,在2.0-4.6V間循環沒有明顯的容量衰減,作者認為是一種面缺陷結構[52]。
總之,富Li和缺Li的層狀化合物均有較好的性能,是值得關注的課題。Zn、Mg等較大離子半徑取代Mn,在Mn3+的價帶引入空穴,產生干擾反鐵磁自旋排列的Mn4+,提高了m-LiMnO2結構的穩定性。Co、Cr、Ni等過渡金屬離子半徑與Mn3+相近,并占據八面體位置,抑制Jahn-Tellen效應,穩定了層狀結構。特別是Ni在層狀中取代,有其良好的性能[53]。
LiMnO2的包覆,與LiMn2O4的包覆機理類似。主要是抑制錳的分解,電解液對電極的侵蝕。金屬氧化物的包覆,金屬M-O的強度,抑制表面氧的活性或提高Li+擴散途徑以提高電化學性能等。
2.6.3 層狀Li2MnO3的復合
Li2MnO3屬巖鹽型單斜晶系結構,空間群為C2/m,與LiMnO2不同的是氧不是理想的密堆積,八面體層中交疊的是Li+與Mn4+層。由于Mn是4價,所以Li2MnO3本身不具電化學活性,Li+也不能脫出,要使之成為正極活性物,方法之一是用3d金屬取代部分Mn,使之形成固溶體復合物,而終端產物可視為Li2MnO3與其他層狀物的復合物。
層狀Li2MnO3可與層狀化合物(LiMO2(M=Co、Ni、Fe……等)形成復合物。由于許多LiMO2與Li2MnO3多為同構物,因而研究過的有(LiCoO2-Li2MnO3,Li1-xNi1-xO2-Li2MnO3,LiCoO2-LiNi0.5Mn0.5O2復合物[54]。這些復合物正極一般都有高電位,高能量密度的特性,因而引起人們的關注。但制備方法和條件都十分苛刻,含Co等成本也昂貴。
LiFeO2也是層狀化合物,LiFeO2-Li2MnO3復合,Fe、Mn均價廉,因而更吸引人,但立方α-LiFeO2與四方γ-LiFeO2和Li2MnO3都是非電化學活性物,要用Fe部分取代Li2MnO3中的Mn就必須用軟化學(低溫)法,即H+/Li+交換或Na+/Li+交換制備法,并限定Fe/Mn+Fe之比。早先用Fe取代Li2MnO3的部分Mn,用固相反應法(Fe/(Fe+Mn)<0.3)或水熱燒結法(Fe/(Fe+ Mn)<0.75)(650℃
由于尖晶石型結構是三維網狀,可以設想與層狀Li2MnO3的復合物的制備要困難一些,已經研究的是Li2MnO3與LiMn2O4的復合,制備出xLi2MnO3 (1-x)Li1+δMn2-δO4 (0
通過H+與Li+間的離子交換,用H2SO4自Li2MnO3層間移去Li2O[30],首先得到的產物是(1-x)[Li(Li0.33Mn0.67)O2] x(0.67MnO2)或(1-x)Li2MnO3 xMnO2,在聚合物鋰電池中以C/15倍率,在2.0-3.8V間充放循環12次后放電容量為155mAh/g。放電曲線表明循環過程中未轉化為尖晶石結構,似逐漸轉化為無定型;隨后嵌入的Li+不能進入Li2MnO3,而只能進入MnO2成為LiMnO2,構成xLi2MnO3 (1-x)LiMnO2。由此也可設想形成(1-x)Li2M′O3 xLiMnO2(M′=Ti4+, M=Ni3+、Mn3+)的新型層狀與層狀結構的復合物,如0.05Li2TiO3 0.95LiMn0.5Ni0.5O2等。
3 結論
錳資源豐富、廉價低廉,對環境友好,安全性又佳,結構研究較為深入,既可用于一次電池,又可用于二次電池。各種晶型之間又可互相轉化,Li-Mn-O化合物-層狀的LiMnO2和三維尖晶石型LiMn2O4是動力電池的正極材料的候選者。相比LiCoO2,Mn-基材料既有優勢,又有發展潛力。
參考文獻
[1] 夏熙. 二氧化錳及相關錳氧化物的晶體結構、制備及放電性能[J]. 電池, 2005, 35(3): 199-203.
[2] 李同慶. 提高堿錳電池性能的研究進展[J]. 電池, 2002, 32(6): 329-333.
[3] 夏熙. 大電流放電堿錳電池的進展[J]. 電池, 2003, 33(2): 83-86.
[4] 王金良, 陳來茂, 陳永心. 鋅錳電池產業現狀與發展方向探討[J]. 電源技術, 2006, 30(2): 89-92.
[5] 馬扣祥, 孟良榮, 夏熙. 高功率放電的二氧化錳和電池[J]. 電源技術, 2007, 31(1): 4-8.
[6] 夏熙. γ-MnO2結構模型現狀與EMD的性能[J]. 電池工業, 2002, 7(3-4): 169-173.